Résumé rapide : L'apprentissage automatique en génomique utilise des algorithmes informatiques pour analyser de vastes ensembles de données génétiques et identifier des schémas invisibles aux méthodes traditionnelles. De la prédiction du risque de maladie à la personnalisation des traitements, les outils d'apprentissage automatique tels que les réseaux neuronaux convolutifs et les modèles d'apprentissage supervisé transforment les données génomiques brutes en informations cliniques, avec des gains de performance de 7 à 291 % par rapport aux approches conventionnelles dans des applications critiques.
Le domaine de la génomique génère plus de données que jamais auparavant. Un seul séquençage de génome entier produit des centaines de gigaoctets. Les méthodes statistiques traditionnelles ne peuvent suivre le rythme.
L'apprentissage automatique change la donne. Les algorithmes entraînés sur des millions de variants génétiques peuvent repérer des schémas qui échapperaient à l'œil humain, prédire les risques de maladie à partir des séquences d'ADN et orienter les décisions thérapeutiques avec une précision sans précédent.
Selon l'Institut national de recherche sur le génome humain (NHGRI), les chercheurs ont de plus en plus recours à l'intelligence artificielle et à l'apprentissage automatique pour identifier des tendances significatives dans des ensembles de données génomiques complexes, à des fins de santé et de recherche. Ce changement n'est pas théorique : il est déjà une réalité dans les cliniques et les laboratoires.
Pourquoi l'apprentissage automatique est important pour la génomique
Les données génomiques sont multidimensionnelles, bruitées et structurées de manière à complexifier les analyses conventionnelles. Un exome typique contient des variants dans des milliers de gènes. Le séquençage de l'exome entier (WES) cible environ 3% du génome, qui constituent la base des gènes codant pour les protéines ; or, même ces 3% génèrent d'énormes ensembles de données présentant les caractéristiques du big data.
L'apprentissage automatique excelle précisément dans ces conditions. Là où les tests statistiques traditionnels peinent à gérer des milliers de variables corrélées, les algorithmes d'apprentissage automatique excellent dans :
- Identification des relations non linéaires entre les variants génétiques et les phénotypes
- Gestion des données manquantes et du bruit technique inhérent au séquençage
- Intégration de sources de données hétérogènes (génomiques, transcriptomiques, cliniques)
- Passage à l'échelle de jeux de données contenant des millions d'échantillons
Ce domaine continue d'étendre l'utilisation des méthodes informatiques pour améliorer la compréhension des schémas cachés dans les grands ensembles de données génomiques complexes, de la recherche fondamentale à la translation clinique.
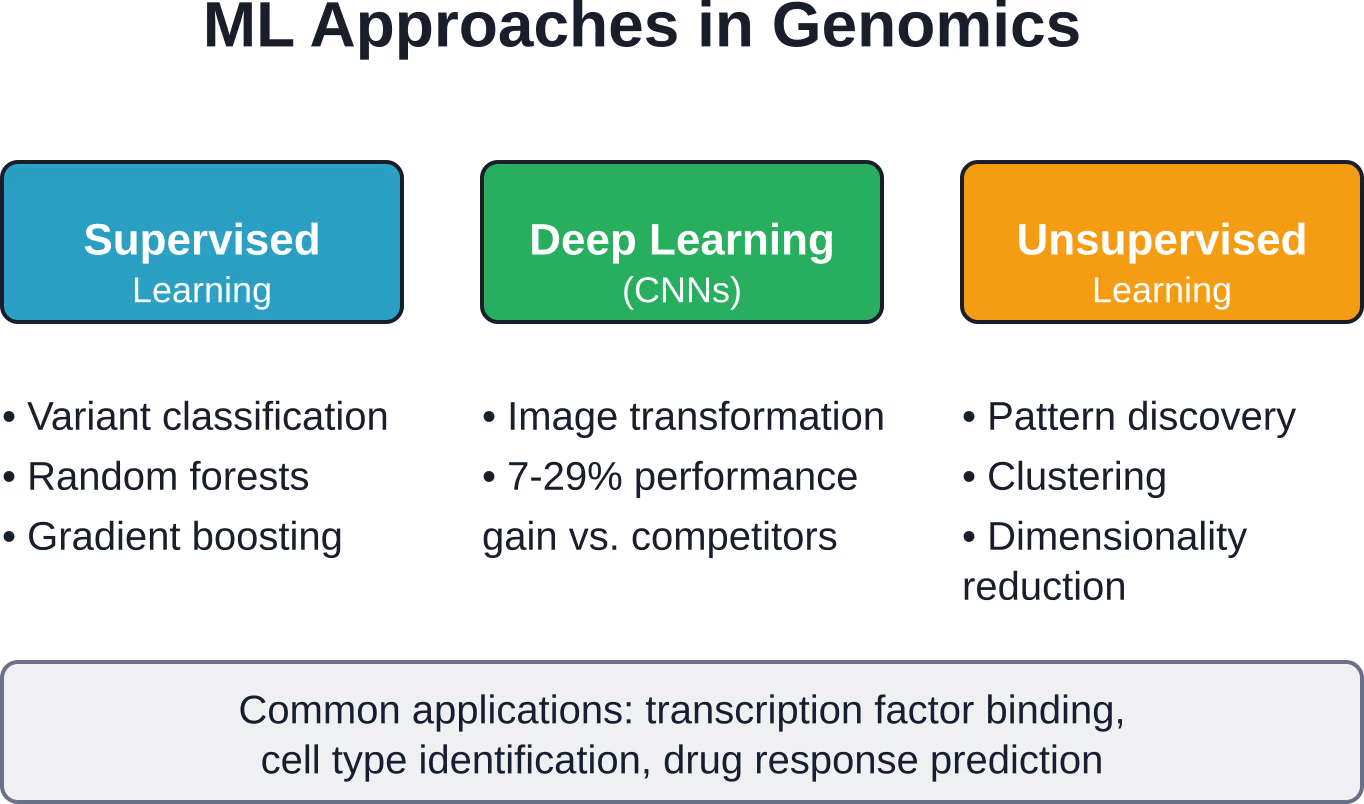
Approches fondamentales d'apprentissage automatique en génomique
Tous les systèmes d'apprentissage automatique ne se valent pas. Différentes questions génomiques exigent différentes stratégies algorithmiques.
Apprentissage supervisé pour la classification des variants
L'apprentissage supervisé utilise des données d'entraînement étiquetées pour construire des modèles prédictifs. En génomique, cela se traduit par l'entraînement d'algorithmes sur des variants pathogènes et bénins connus afin de classifier de nouveaux variants incertains.
Les techniques supervisées courantes comprennent :
- Forêts aléatoires combinant arbres de décision pour prédire la pathogénicité des variants
- Machines à vecteurs de support qui trouvent les frontières optimales entre les classes de variants
- Méthodes de gradient boosting qui affinent itérativement les prédictions
Ces méthodes alimentent les bases de données de variants cliniques et les outils de prédiction utilisés quotidiennement dans les laboratoires de diagnostic. La médecine génomique, qui permet d'établir des diagnostics et de prendre des décisions thérapeutiques en fonction des variations génomiques, est désormais intégrée à la pratique clinique et plus accessible. L'interprétation clinique des variations génomiques détectées par l'analyse du génome est cruciale en médecine génomique.
Apprentissage profond et réseaux neuronaux convolutifs
L'apprentissage profond représente un changement révolutionnaire dans la modélisation prédictive grâce à l'application de réseaux neuronaux multicouches, en particulier de réseaux neuronaux convolutifs (CNN).
Or, il faut savoir que les CNN ont été initialement conçus pour l'analyse d'images. Les chercheurs ont développé des méthodes de transformation comme DeepInsight qui convertissent les données génomiques de leur format tabulaire en représentations de type image, permettant ainsi aux CNN de capturer efficacement les caractéristiques latentes.
Les résultats sont éloquents. D'après une étude publiée dans Nature, DeepInsight-3D a démontré une amélioration des performances (mesurée par l'aire sous la courbe ROC) de 7 à 291 TP3T par rapport à l'ensemble des méthodes testées. DeepInsight-3D a atteint une aire sous la courbe moyenne de 0,72 pour la prédiction de la réponse au médicament.
L'apprentissage par transfert réduit encore le temps de calcul et améliore les performances. Les modèles pré-entraînés sur de grands ensembles de données génomiques peuvent être affinés pour des tâches spécifiques à l'aide d'ensembles de données plus petits, améliorant ainsi les performances dans des tâches telles que la prédiction de la liaison des facteurs de transcription.
Apprentissage non supervisé pour la découverte de modèles
En l'absence de données d'entraînement étiquetées, l'apprentissage non supervisé découvre la structure des données génomiques sans catégories prédéfinies.
Ces techniques comprennent des algorithmes de clustering qui regroupent les échantillons similaires et des méthodes de réduction de dimensionnalité qui visualisent les données génomiques de haute dimension en deux ou trois dimensions. Ces approches révèlent des structures de population cachées, identifient des sous-types de maladies et suggèrent de nouvelles hypothèses biologiques.

Appliquer l'apprentissage automatique à la recherche en génomique grâce à l'IA supérieure
L'apprentissage automatique remodèle la génomique en aidant les chercheurs à analyser de vastes ensembles de données génétiques et à découvrir des tendances significatives. IA supérieure propose des solutions d'IA et d'apprentissage automatique personnalisées qui peuvent être appliquées aux défis complexes liés aux données dans la recherche en génomique.
Intégrez l'IA à vos flux de travail en génomique
AI Superior offre des capacités d'apprentissage automatique qui peuvent soutenir des initiatives en génomique, telles que :
- Reconnaissance de formes dans les données à grande échelle
- Des modèles prédictifs pour aider à identifier les tendances
- Automatisation du traitement des données et des flux de travail analytiques
👉Contactez l'IA supérieure Contactez-nous aujourd'hui pour découvrir comment leur expertise en IA peut soutenir vos recherches en génomique.
Des applications concrètes transforment la recherche et les soins
L'apprentissage automatique en génomique ne se limite pas aux articles scientifiques. Ses applications transforment la pratique clinique et la recherche biologique.
Prédiction de la pathogénicité des variants
Les bases de données de variants cliniques et les algorithmes de prédiction par apprentissage automatique aident les cliniciens à interpréter les milliers de variants découverts dans les génomes des patients. Les outils entraînés sur des bases de données comme ClinVar et COSMIC permettent de prédire si les variants nouvellement découverts sont susceptibles d'être à l'origine d'une maladie.
Ces prédictions orientent les décisions diagnostiques, le dépistage familial et le choix du traitement dans les maladies génétiques rares et le cancer.
Réponse aux médicaments et oncologie de précision
Les bases de données multi-omiques du cancer, associées à des modèles d'apprentissage automatique, permettent de prédire la réponse des tumeurs à des thérapies spécifiques. En analysant conjointement les données génomiques, transcriptomiques et protéomiques, les algorithmes identifient les patients les plus susceptibles de bénéficier de traitements ciblés.
L’encyclopédie des lignées cellulaires cancéreuses (CCLE), la base de données sur la génomique de la sensibilité aux médicaments dans le cancer (GDSC) et l’Atlas du génome du cancer (TCGA) fournissent des données d’entraînement pour ces modèles. Les chercheurs ont atteint une précision de 721 % (TP3T) dans la prédiction de l’efficacité des médicaments grâce à des techniques d’apprentissage profond appliquées à ces ensembles de données.
Liaison des facteurs de transcription et régulation des gènes
Comprendre où les facteurs de transcription se lient à l'ADN est fondamental pour décrypter la régulation des gènes. Les modèles d'apprentissage automatique entraînés sur des données ChIP-seq et DNase-seq prédisent les sites de liaison à partir de la seule séquence d'ADN.
Le dépôt Kipoi accélère l'échange et la réutilisation de modèles prédictifs en génomique au sein de la communauté, en hébergeant des modèles de liaison des facteurs de transcription, d'épissage de l'ARN et d'accessibilité de la chromatine. Cette approche collaborative évite le développement de modèles redondants et permet une évaluation comparative systématique.
Identification du type cellulaire à partir de données unicellulaires
Le séquençage d'ARN unicellulaire génère des profils d'expression pour des milliers de cellules individuelles. L'apprentissage automatique automatise la classification des types cellulaires, remplaçant l'annotation manuelle par des algorithmes évolutifs et reproductibles.
D'après des recherches publiées, les méthodes utilisant des approches d'apprentissage profond ont montré des améliorations de performance pour l'identification des types cellulaires.
| Domaine d'application | Méthode ML | Indicateur de performance | Impact clinique |
|---|---|---|---|
| pathogénicité variante | Forêts aléatoires, SVM | AUC 0,85-0,95 | Classification diagnostique |
| sites de liaison du TF | CNN | Gain AUPRC 15.1% | Compréhension réglementaire |
| ID du type de cellule | scDeepInsight | Amélioration 7% | sous-typage des maladies |
Ingénierie des caractéristiques génomiques et entrées du modèle
Le succès des modèles d'apprentissage automatique dépend crucialement de la manière dont les données génomiques sont représentées et des caractéristiques qui sont extraites.
Des analyses génomiques à grande échelle ont révélé des schémas prédictifs associés aux caractéristiques et aux modes de vie des organismes. Une étude portant sur 387 génomes fongiques a utilisé des ensembles de caractéristiques dérivés d'enzymes actives sur les glucides (CAZymes), de peptidases, de groupes de métabolites secondaires, de transporteurs et de facteurs de transcription.
Bien que la phylogénie ait constitué un élément important de la plupart des prédictions, l'intégration de données génomiques a amélioré les performances de prédiction pour chaque mode de vie et caractéristique testés. Pour la prédiction du mode de vie biotrophe obligatoire, les données phylogénétiques seules ont permis d'obtenir une AUC de 0,899 ± 0,018, mais l'ajout de caractéristiques génomiques a porté ces performances à 1,000 ± 0,000, démontrant ainsi une amélioration substantielle grâce à l'intégration de ces caractéristiques.
Soyons francs : le choix des fonctionnalités fait souvent la différence entre un modèle médiocre et un modèle révolutionnaire.
Principales catégories de fonctionnalités
- Caractéristiques basées sur les séquences : Fréquences des k-mers, teneur en GC, occurrences de motifs
- Annotations fonctionnelles : Termes d'ontologie génique, appartenance à des voies métaboliques, domaines protéiques
- Caractéristiques évolutives : Scores de conservation, signaux phylogénétiques, relations d'homologie
- Caractéristiques structurelles : Prédictions de structure secondaire, état de la chromatine, forme de l'ADN
Pour la prédiction des nécrotrophes dans les génomes fongiques, l'augmentation maximale du score AUC était de 0,395 en utilisant l'ensemble de caractéristiques CAZyme, soit un gain AUC moyen de 87% sur les trois meilleurs ensembles de caractéristiques par rapport aux méthodes de parcimonie.
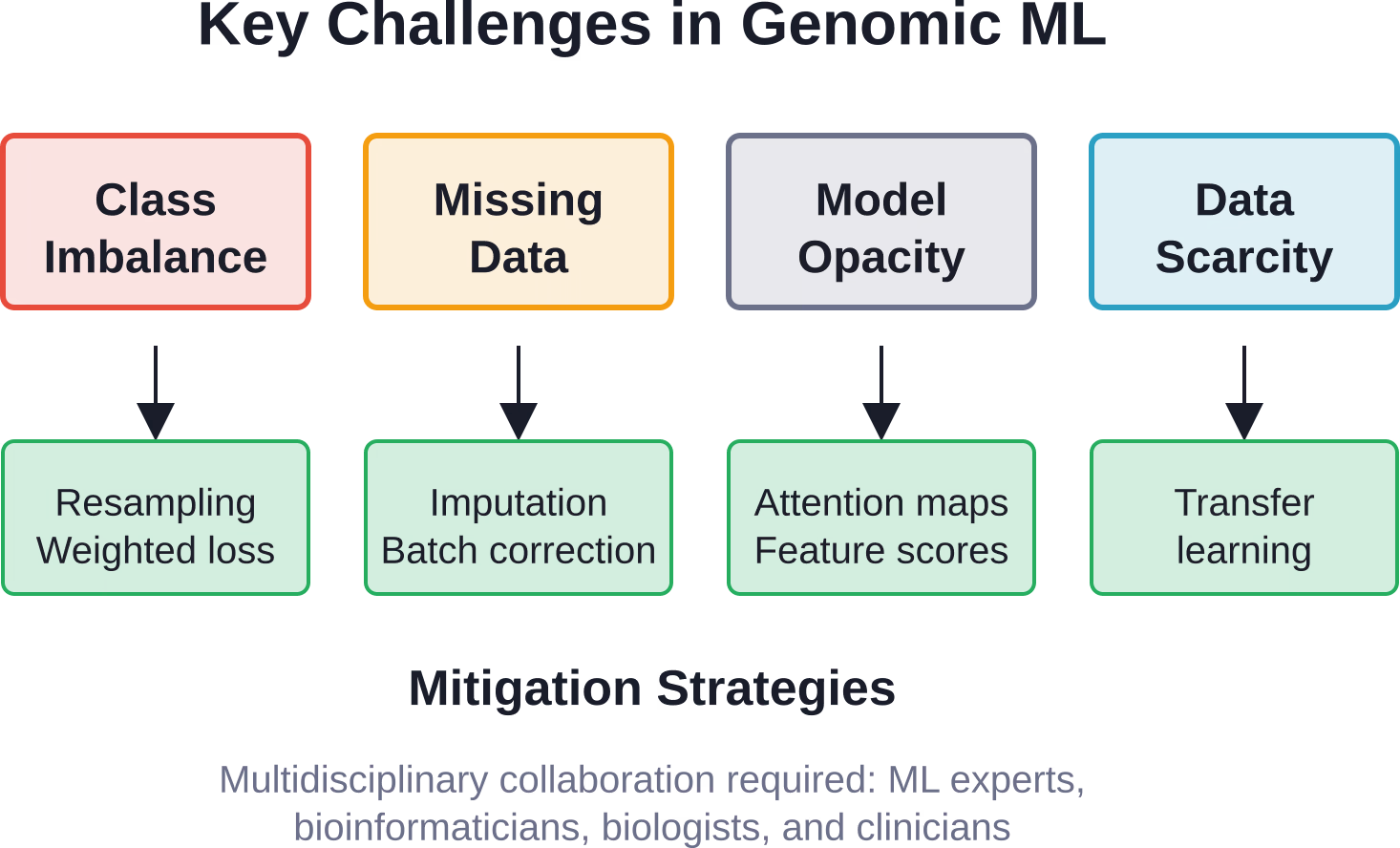
Défis que l'apprentissage automatique doit surmonter
Malgré des succès impressionnants, l'apprentissage automatique en génomique se heurte à de réels obstacles qui limitent ses applications actuelles.
Des classes de tailles déséquilibrées
Les variants pathogènes sont rares comparés aux variants bénins. Les cas de maladie sont moins nombreux que les témoins. Ce déséquilibre biaise les modèles en faveur du variant majoritaire, réduisant ainsi la sensibilité aux événements rares qui ont une importance clinique majeure.
Les solutions comprennent des techniques de rééchantillonnage, des fonctions de perte pondérées et des méthodes d'ensemble qui traitent explicitement le déséquilibre.
Données manquantes et hétérogènes
Les jeux de données génomiques contiennent fréquemment des valeurs manquantes dues à des défaillances techniques, à une absence biologique ou à des bases de données incomplètes. Les différentes plateformes de séquençage, les protocoles et les chaînes de traitement introduisent des effets de lot et une hétérogénéité.
Les méthodes d'imputation avancées et les techniques d'adaptation de domaine sont utiles, mais le traitement des données hétérogènes reste un domaine de recherche actif.
Interprétabilité du modèle
Les modèles d'apprentissage profond sont souvent des “ boîtes noires ”. Un réseau neuronal peut prédire avec précision le risque de maladie, mais ne fournit aucune explication mécanistique sur les raisons pour lesquelles une variante est pathogène.
Pour une application clinique, l'interprétabilité est cruciale. Des techniques comme les mécanismes d'attention, les cartes de saillance et les scores d'importance des caractéristiques offrent des solutions partielles, révélant quelles régions génomiques influencent les prédictions.
Taille et qualité des données
L'apprentissage automatique est très gourmand en données. L'entraînement de modèles robustes nécessite des milliers, voire des millions, d'exemples étiquetés. Pour les maladies rares ou les populations peu étudiées, ces données n'existent tout simplement pas encore.
Les approches d'apprentissage par transfert et d'apprentissage avec peu d'exemples visent à construire des modèles utiles à partir de données limitées, mais la rareté des données reste une contrainte fondamentale.
Outils et ressources pour accélérer le développement
L'écosystème de la génomique et de l'apprentissage automatique comprend des référentiels, des bases de données et des cadres collaboratifs qui réduisent les barrières à l'entrée.
Référentiels de modèles
Le dépôt Kipoi héberge des modèles pré-entraînés pour les applications en génomique, permettant aux chercheurs d'utiliser des modèles existants sans avoir à les réentraîner. Cela accélère les échanges et la réutilisation des modèles prédictifs au sein de la communauté.
Parmi les autres dépôts, on trouve :
- MLOmics : Base de données multi-omiques sur le cancer, spécifiquement structurée pour les applications d'apprentissage automatique
- Collections GitHub : Dépôts de code maintenus par la communauté pour les flux de travail d'apprentissage automatique en génomique
Initiatives gouvernementales et institutionnelles
L’Institut national de recherche sur le génome humain (NHGRI) a créé le consortium MAGen (ML/AI Tools to Advance Genomic Translational Research). Ce projet de recherche collaborative explore la faisabilité d’outils d’apprentissage automatique et d’intelligence artificielle susceptibles d’améliorer la précision des prédictions concernant la manière dont les individus porteurs de variants génétiques pathogènes manifestent la maladie.
MAGen réunit le National Institute on Aging (NIA), l'Office of Data Science and Strategy (ODSS) et le NHGRI pour répondre aux questions critiques de la recherche translationnelle en génomique grâce au développement coordonné de l'apprentissage automatique.
Ressources pédagogiques
Des cours et des tutoriels aident les chercheurs à acquérir les compétences informatiques nécessaires pour appliquer l'apprentissage automatique aux problèmes de génomique. Des plateformes en ligne proposent des cours spécialisés en apprentissage automatique appliqué à la génomique, tandis que les programmes universitaires intègrent de plus en plus la génomique computationnelle dans leurs cursus.
L'avenir de l'apprentissage automatique en génomique
Quelle sera l'évolution de ce secteur à partir de maintenant ? Plusieurs tendances se dessinent.
Intégration multimodale
La prochaine génération de modèles intégrera les séquences génomiques aux données transcriptomiques, protéomiques, métabolomiques et cliniques. Les approches multi-omiques permettent de saisir la complexité du vivant de manière plus complète que les données prises individuellement.
Les premiers résultats sont prometteurs. Les modèles combinant données génomiques et transcriptomiques surpassent les approches unimodales dans de multiples tâches de prédiction.
Modèles fondamentaux pour la génomique
Les grands modèles de langage ont transformé le traitement automatique du langage naturel. Les modèles génomiques de base — des réseaux neuronaux massifs pré-entraînés sur des milliards de séquences d'ADN et d'ARN — commencent à montrer un potentiel similaire.
Ces modèles apprennent les schémas fondamentaux de la biologie du génome lors du pré-entraînement, puis s'adaptent rapidement à des tâches spécifiques avec un minimum de données d'ajustement. Cette approche pourrait démocratiser l'apprentissage automatique génomique en réduisant les besoins en données pour le développement de modèles fonctionnels.
Méthodes de préservation de la vie privée
Les données génomiques sont intrinsèquement sensibles et identifiables. L'apprentissage fédéré permet l'entraînement de modèles dans plusieurs institutions sans centralisation des données brutes. La confidentialité différentielle offre des garanties mathématiques supplémentaires empêchant la divulgation d'informations individuelles dans les résultats des modèles.
Ces techniques seront essentielles lorsque la médecine génomique s'appliquera à l'échelle de la population.
Aide à la décision clinique
Les outils d'apprentissage automatique passent du stade de prototypes de recherche à celui de systèmes d'aide à la décision clinique homologués par la FDA. Il faut s'attendre à une croissance continue des procédures réglementaires pour l'IA génomique, à la normalisation des critères de performance et à l'intégration aux dossiers médicaux électroniques.
Mais attention ! L’adoption clinique ne se limite pas aux performances techniques. L’interprétabilité, la réduction des biais et les considérations d’équité détermineront si ces outils améliorent ou aggravent les inégalités en matière de santé.
Premiers pas avec l'apprentissage automatique génomique
Pour les chercheurs souhaitant appliquer l'apprentissage automatique aux problèmes de génomique, plusieurs étapes pratiques permettent d'acquérir des compétences fondamentales :
- Apprenez la biologie : Un apprentissage automatique génomique efficace nécessite la compréhension des questions biologiques et des processus de génération de données.
- Maîtriser les techniques fondamentales du ML : Commencez par les principes fondamentaux de l'apprentissage supervisé avant de passer à l'apprentissage en profondeur.
- Explorez les ensembles de données publics : TCGA, CCLE, GDSC, ClinVar et gnomAD fournissent des données d'entraînement pour diverses applications.
- Utiliser des cadres établis : Les bibliothèques Python telles que scikit-learn, TensorFlow et PyTorch accélèrent le développement
- Évaluer rigoureusement : Comparer les nouvelles méthodes aux méthodes de référence établies à l'aide d'ensembles de test mis de côté
- Collaborer entre les disciplines : Collaborer avec des experts du domaine pour garantir la pertinence biologique et l'utilité clinique
Les ressources gérées par la communauté, comme les dépôts GitHub et les cours en ligne, facilitent l'apprentissage. Le domaine bénéficie d'une culture open source et du partage de données qui permettent une itération rapide.
Questions fréquemment posées
Qu'est-ce que l'apprentissage automatique en génomique ?
L'apprentissage automatique en génomique utilise des algorithmes informatiques pour analyser les données génétiques, identifier des schémas et prédire les fonctions biologiques, les risques de maladies et la réponse aux traitements. Ces méthodes permettent de gérer plus efficacement la nature complexe et multidimensionnelle des ensembles de données génomiques que les approches statistiques traditionnelles.
Dans quelle mesure les modèles d'apprentissage automatique sont-ils précis pour la prédiction génomique ?
La précision varie selon l'application. Les classificateurs de pathogénicité des variants atteignent des scores AUC de 0,85 à 0,95. Les méthodes DeepInsight présentent des améliorations de performance de 7 à 29% par rapport aux approches concurrentes. Les performances dépendent de la qualité des données d'entraînement, de l'ingénierie des caractéristiques et de la tâche de prédiction spécifique.
Quels sont les principaux défis liés à l'application du ML à la génomique ?
Les principaux défis comprennent le déséquilibre entre les variants rares et communs, les données manquantes ou hétérogènes issues de différentes plateformes de séquençage, l'interprétabilité des modèles pour la prise de décision clinique et la quantité limitée de données d'entraînement pour les maladies rares ou les populations sous-représentées. Pour y remédier, une collaboration multidisciplinaire entre experts en apprentissage automatique, bioinformaticiens et cliniciens est indispensable.
L'apprentissage automatique peut-il prédire les maladies à partir de séquences d'ADN ?
Les modèles d'apprentissage automatique peuvent estimer le risque de maladie à partir de variants génomiques, mais leurs prédictions sont probabilistes et non déterministes. Les modèles entraînés sur de vastes bases de données comme ClinVar prédisent la pathogénicité des variants afin d'orienter le diagnostic. Les scores de risque polygénique combinent les effets de nombreux variants pour estimer la susceptibilité à la maladie. Cependant, les facteurs environnementaux, les interactions gène-environnement et les connaissances biologiques incomplètes limitent la précision des prédictions.
Quelle est la différence entre l'apprentissage supervisé et l'apprentissage non supervisé en génomique ?
L'apprentissage supervisé utilise des données d'entraînement étiquetées (par exemple, des variants marqués comme pathogènes ou bénins) pour construire des modèles prédictifs. Il est utilisé pour les tâches de classification et de régression. L'apprentissage non supervisé découvre des structures dans des données non étiquetées grâce au clustering et à la réduction de dimensionnalité, révélant ainsi la structure des populations ou les sous-types de maladies sans catégories prédéfinies.
Comment l'apprentissage profond améliore-t-il l'analyse génomique ?
L'apprentissage profond, et notamment les réseaux de neurones convolutifs, apprend automatiquement les caractéristiques hiérarchiques à partir de données brutes. Des méthodes comme DeepInsight transforment les données génomiques tabulaires en représentations visuelles, permettant ainsi aux CNN de capturer des relations non linéaires complexes. L'apprentissage par transfert permet d'affiner des modèles pré-entraînés sur de vastes ensembles de données pour des tâches spécifiques, améliorant ainsi les performances avec moins de données et de puissance de calcul.
Quelles sont les ressources disponibles pour apprendre l'apprentissage automatique en génomique ?
Le dépôt Kipoi héberge des modèles pré-entraînés et du code. Le consortium MAGen du NHGRI développe des outils collaboratifs d'apprentissage automatique. Des cours en ligne enseignent les fondamentaux de l'apprentissage automatique en génomique. Des bases de données publiques (TCGA, CCLE, GDSC, ClinVar) fournissent des données d'entraînement. Les bibliothèques Python (scikit-learn, TensorFlow, PyTorch) offrent des cadres d'implémentation. Les dépôts GitHub partagent des flux de travail et des tutoriels développés par la communauté.
Conclusion
L'apprentissage automatique transforme radicalement la manière dont les chercheurs et les cliniciens interprètent les données génomiques. De la prédiction de la pathogénicité des variants à la personnalisation des traitements anticancéreux, les algorithmes d'apprentissage automatique offrent des perspectives impossibles à obtenir par les méthodes traditionnelles.
Les gains de performance sont quantifiables : amélioration de la précision du modèle (7–29%) et obtention de scores AUC parfaits pour certaines tâches de classification. Il ne s’agit pas de progrès incrémentaux, mais de véritables avancées.
Des défis subsistent. La rareté des données, l'interprétabilité des modèles et l'accès équitable exigent une attention constante. Mais la tendance est claire : l'apprentissage automatique deviendra aussi fondamental en génomique que le séquençage lui-même.
Pour les chercheurs, il est urgent de développer des compétences en apprentissage automatique. Pour les cliniciens, la maîtrise de ces outils est de plus en plus essentielle à une pratique fondée sur les preuves. Le domaine de la génomique continue d'enrichir les méthodes informatiques afin de mieux comprendre les schémas cachés – et ces schémas commencent à peine à se révéler.
Prêt à explorer l'apprentissage automatique dans vos recherches en génomique ? Commencez par des ensembles de données publics, tirez parti des modèles pré-entraînés provenant de référentiels comme Kipoi et collaborez avec des experts en informatique pour garantir la pertinence biologique et l'impact clinique.