Résumé rapide : L'apprentissage automatique révolutionne la découverte de médicaments en accélérant le criblage moléculaire, en prédisant les interactions médicament-cible et en optimisant les propriétés chimiques. Cette technologie répond au principal défi de l'industrie : le développement traditionnel d'un médicament prend plus de dix ans et coûte en moyenne 1 400 milliards de dollars, avec un taux de réussite d'environ 6,21 % entre les essais de phase I et l'autorisation de mise sur le marché. Les modèles d'apprentissage automatique aident désormais les entreprises pharmaceutiques à identifier plus rapidement les composés prometteurs, à prédire plus tôt leur toxicité et à réduire les échecs coûteux en phase finale de développement.
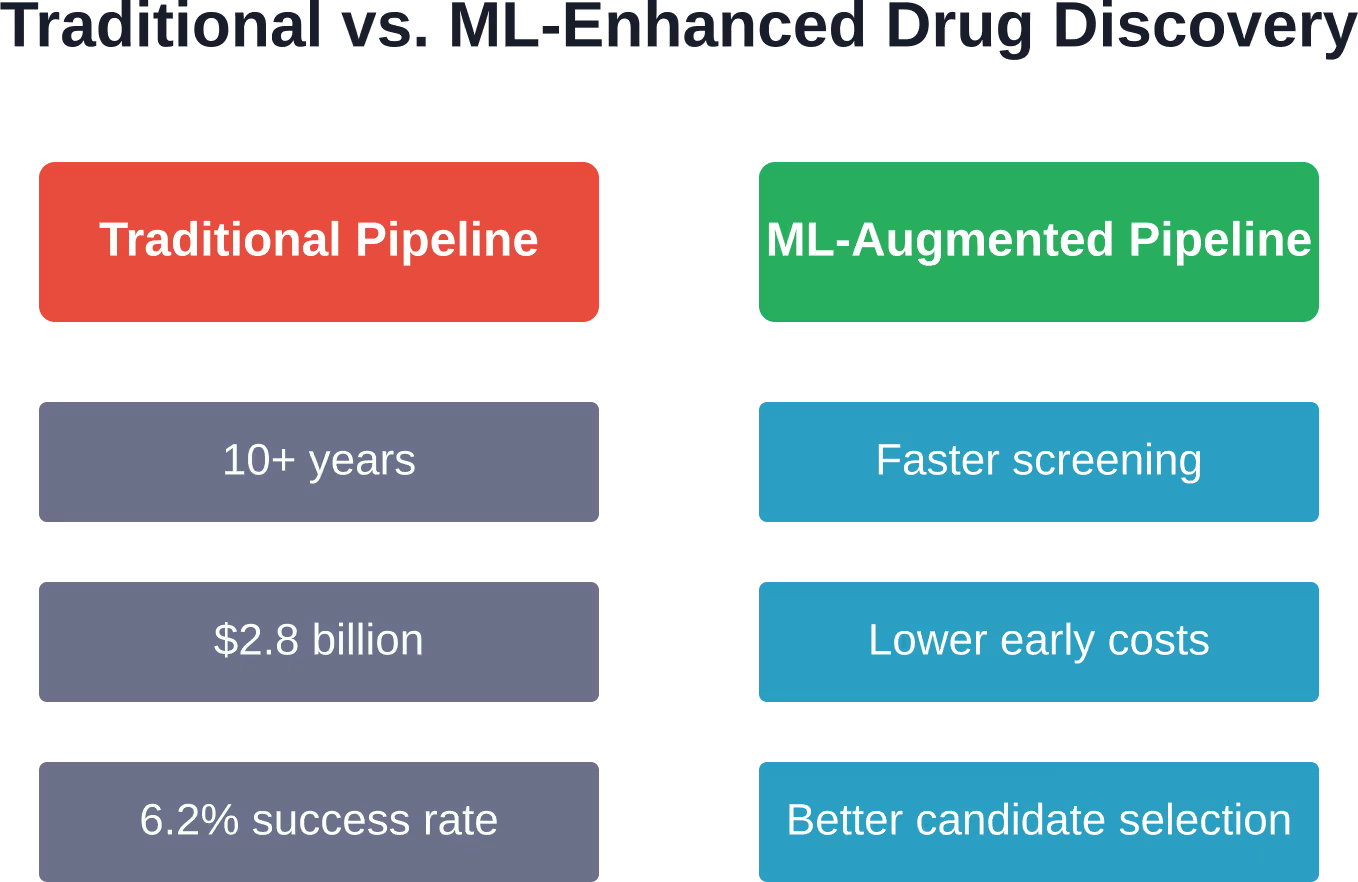
L'industrie pharmaceutique est confrontée à une dure réalité. Le développement d'un seul médicament prend plus de dix ans et coûte en moyenne 1 400 milliards de dollars américains, selon les études médicales. Malgré cet investissement colossal, 9 molécules thérapeutiques sur 10 échouent entre les essais cliniques de phase II et l'autorisation de mise sur le marché.
L'apprentissage automatique s'est imposé comme un outil puissant pour remédier à ces inefficacités considérables. En analysant de vastes chimiothèques, en prédisant le comportement moléculaire et en identifiant plus tôt les candidats médicaments prometteurs, les techniques d'apprentissage automatique transforment en profondeur la manière dont les chercheurs abordent la découverte de médicaments.
Le défi de la découverte de médicaments
Le développement traditionnel des médicaments suit un processus linéaire et long. Les scientifiques examinent des milliers de composés expérimentalement, les testent sur des cultures cellulaires, évaluent les candidats prometteurs sur des modèles animaux, et ce n'est qu'ensuite qu'ils passent aux essais cliniques chez l'humain. Chaque étape exige des années de travail et des millions d'euros de financement.
Les chiffres sont alarmants. Sur 21 143 composés étudiés, le taux de réussite global, des essais cliniques de phase I à l’autorisation de mise sur le marché, est d’environ 6,21 %. Autrement dit, sur 100 médicaments testés chez l’humain, moins de sept sont commercialisés.
Comment l'apprentissage automatique change la donne
L'apprentissage automatique introduit une approche fondamentalement différente. Au lieu de tester les composés un par un en laboratoire, les modèles d'apprentissage automatique peuvent évaluer par calcul des millions de structures moléculaires, prédisant leurs chances de succès avant même le début d'une seule expérience.
Cette technologie excelle dans la détection de motifs au sein de données chimiques et biologiques multidimensionnelles, des motifs imperceptibles à l'œil nu pour les chercheurs. Un réseau neuronal peut analyser la structure tridimensionnelle d'une protéine, prédire comment des milliers de petites molécules pourraient s'y lier et classer les candidats selon leur efficacité prédite.

Appliquer l'apprentissage automatique à la découverte de médicaments grâce à l'IA supérieure
L'apprentissage automatique est utilisé pour traiter de vastes ensembles de données biologiques et chimiques et pour faciliter la prise de décision dans la recherche en phase préliminaire. IA supérieure propose des services de conseil en IA et de développement de systèmes d'apprentissage automatique personnalisés pour les applications axées sur les données dans le secteur de la santé et les domaines connexes.
Besoin d'aide pour créer une solution d'apprentissage automatique pour la découverte de médicaments ?
AI Superior prend en charge :
- Développement de modèles d'apprentissage automatique personnalisés
- Analyse des données et modélisation prédictive
- Solutions de vision par ordinateur et de reconnaissance de formes
- Conseil en IA et développement de preuves de concept
👉Contactez l'IA supérieure pour discuter de votre projet d'apprentissage automatique pour la découverte de médicaments.
Principales applications du ML tout au long du pipeline
Dépistage virtuel et découverte de succès
La première étape de la découverte de médicaments consiste à identifier des molécules actives, c'est-à-dire des molécules présentant une activité biologique quelconque contre une cible pathologique. Traditionnellement, cela impliquait de tester physiquement des dizaines de milliers de composés en laboratoire.
Le criblage virtuel basé sur l'apprentissage automatique bouleverse ce modèle. Des algorithmes d'apprentissage profond, entraînés sur des bases de données de structures chimiques, peuvent prédire quelles molécules sont les plus susceptibles de se lier à une protéine cible spécifique. Les chercheurs ne testent ensuite expérimentalement que les candidats les plus prometteurs, ce qui réduit considérablement le nombre de composés à synthétiser et à tester.
Prédiction des interactions médicament-cible
Comprendre comment une petite molécule interagit avec sa cible biologique est essentiel au développement de médicaments. Se lie-t-elle suffisamment fortement ? Active-t-elle ou inhibe-t-elle la protéine cible ? Provoquera-t-elle des effets indésirables ?
Les modèles d'apprentissage automatique abordent ces questions par de multiples approches. Les réseaux neuronaux graphiques peuvent représenter les molécules et les protéines sous forme de graphes mathématiques, apprenant ainsi à prédire l'affinité de liaison à partir de caractéristiques structurales. Les réseaux neuronaux récurrents avec apprentissage par renforcement affichent d'excellentes performances dans les tâches de notation.
Optimisation immobilière et développement des prospects
Trouver une molécule qui cible un médicament n'est que le point de départ. Cette molécule initiale doit être optimisée pour présenter des propriétés pharmacologiques : biodisponibilité orale, stabilité métabolique, franchissement de la barrière hémato-encéphalique, faible toxicité et facilité de fabrication.
Les modèles d'apprentissage automatique facilitent la navigation dans ce paysage complexe d'optimisation. En s'entraînant sur des ensembles de données reliant les structures chimiques aux propriétés mesurées, les algorithmes apprennent à prédire comment les modifications structurales affecteront le comportement d'un composé. Les chimistes médicinaux peuvent ainsi explorer par calcul des millions de variantes chimiques avant de synthétiser les options les plus prometteuses.
Les arbres de décision et les modèles de forêts aléatoires présentent des niveaux de précision variables dans l'analyse des traitements médicamenteux. Ces méthodes d'ensemble combinent plusieurs arbres de décision pour produire des prédictions robustes, même lorsque les données d'apprentissage sont bruitées ou incomplètes.

Qualité des données : le fondement du succès
La qualité des modèles d'apprentissage automatique dépend de la qualité des données utilisées pour leur entraînement. L'industrie pharmaceutique a consacré des décennies à la production de données biologiques et chimiques, mais une grande partie de ces données se trouve dans des bases de données propriétaires ou des articles publiés, dans des formats qui nécessitent un nettoyage approfondi.
La préparation des données représente la majeure partie des efforts dans tout projet de découverte de médicaments par apprentissage automatique. Les structures chimiques doivent être standardisées. Les mesures expérimentales nécessitent un contrôle qualité afin d'éliminer les valeurs aberrantes et les erreurs. Les données des essais biologiques doivent être normalisées entre les différentes plateformes expérimentales.
Ce travail de prétraitement, souvent ingrat, est déterminant pour la réussite ou l'échec d'un modèle. Un réseau neuronal profond entraîné sur des données bruitées et incohérentes produira des prédictions peu fiables : si les données d'entrée sont erronées, les résultats le seront également. Les équipes qui investissent massivement dans la curation et la validation des données surpassent systématiquement celles qui se concentrent sur les dernières innovations algorithmiques.
Limitations et défis actuels
Malgré des progrès impressionnants, l'apprentissage automatique appliqué à la découverte de médicaments se heurte à des obstacles importants. Les modèles entraînés sur un type d'espace chimique donné échouent souvent lorsqu'ils sont appliqués à des composés structurellement différents. L'apprentissage par transfert apporte des améliorations, mais ne résout pas entièrement le problème de la généralisation.
L'interprétabilité demeure une préoccupation majeure. Lorsqu'un réseau neuronal opaque prédit le succès du composé X et l'échec du composé Y, les chimistes médicinaux cherchent à comprendre pourquoi. Si les techniques d'IA explicable progressent, de nombreux modèles restent encore des oracles impénétrables.
Le secteur est également confronté à des difficultés de validation. Un modèle peut atteindre une précision de 95 % (TP3T) sur des données de test indépendantes, mais cela se traduit-il par un succès concret ? La validation prospective – où les prédictions d’apprentissage automatique sont testées expérimentalement en laboratoire – constitue la preuve ultime, et de nombreux modèles publiés n’ont pas fait l’objet de cet examen rigoureux.
Le paysage réglementaire
La FDA a commencé à publier des recommandations sur l'utilisation de l'IA et de l'apprentissage automatique dans le développement des médicaments. En janvier 2025, elle a publié un projet de recommandations sur l'utilisation de l'intelligence artificielle pour le développement des médicaments et des produits biologiques.
Cette attention réglementaire représente à la fois une opportunité et un défi. D'une part, la reconnaissance de la FDA légitime l'apprentissage automatique comme un outil précieux dans la recherche pharmaceutique. D'autre part, les entreprises doivent désormais démontrer que leurs systèmes d'IA répondent aux normes de transparence, de reproductibilité et de validation – des exigences qui complexifient leur déploiement.
| Domaine d'application de l'apprentissage automatique | Avantage principal | Défi clé |
|---|---|---|
| Projection virtuelle | Tester des millions de composés par calcul | prédictions faussement positives |
| Prédiction de la cible | Identifier de nouveaux liens entre médicaments et maladies | Données d'entraînement limitées pour les maladies rares |
| Optimisation immobilière | Naviguer dans l'espace de conception multi-objectifs | Équilibrer les propriétés concurrentes |
| Prédiction de la toxicité | Signaler rapidement les composés dangereux | Lacunes dans la compréhension des mécanismes de toxicité |
| Conception d'essais cliniques | Stratification des patients et sélection du critère d'évaluation | Contraintes en matière de confidentialité et de partage des données |
Impact concret et études de cas
Les entreprises pharmaceutiques et les jeunes pousses de biotechnologie déploient activement l'apprentissage automatique dans leurs processus de développement. Les principaux instituts de recherche proposent désormais des formations spécialisées en apprentissage automatique appliqué à la découverte de médicaments, témoignant de la maturité du domaine.
La recherche académique repousse sans cesse les limites. L'intégration de l'apprentissage automatique ne se limite plus aux phases préliminaires de la recherche. Les modèles d'apprentissage automatique contribuent désormais à l'optimisation des essais cliniques, au recrutement des patients, à la prédiction des effets indésirables et au contrôle des procédés de fabrication. L'ensemble du cycle de vie du développement des médicaments intègre de plus en plus l'aide à la décision algorithmique.
Perspectives d'avenir : Le pipeline intégrant l'apprentissage automatique
La prochaine génération de découverte de médicaments ne considérera pas l'apprentissage automatique comme une option. Au contraire, il constituera l'épine dorsale informatique de la recherche pharmaceutique, intégré à chaque étape, de l'identification initiale de la cible jusqu'à la surveillance post-commercialisation.
Des approches hybrides émergent déjà, où les prédictions d'apprentissage automatique orientent la conception expérimentale et où les résultats expérimentaux permettent d'améliorer les modèles. Ce cycle itératif accélère l'apprentissage bien au-delà de ce que les humains ou les algorithmes pourraient accomplir seuls.
L'intelligence artificielle générative représente la toute nouvelle frontière. Plutôt que de se contenter de cribler des composés existants, les modèles génératifs peuvent concevoir de nouvelles structures moléculaires optimisées pour des propriétés spécifiques. Ces molécules conçues par l'IA explorent souvent un espace chimique que les chimistes humains n'envisageraient pas intuitivement, menant ainsi à des thérapies véritablement innovantes.
Mais la technologie seule ne suffira pas à relever les défis de la découverte de médicaments. Le succès repose sur la collaboration entre les data scientists, experts en apprentissage automatique, et les spécialistes du domaine, qui maîtrisent la biologie, la chimie et la médecine. Les équipes les plus performantes associent puissance de calcul et connaissances scientifiques approfondies.
Questions fréquemment posées
Dans quelle mesure l'apprentissage automatique permet-il de réduire les coûts de développement des médicaments ?
L'apprentissage automatique peut réduire considérablement les coûts des phases précoces en diminuant le nombre de composés nécessitant une synthèse et des tests physiques. Alors que le processus traditionnel coûte en moyenne 1 à 4 milliards de dollars par médicament approuvé, le criblage optimisé par l'apprentissage automatique permet aux chercheurs de concentrer leurs ressources expérimentales sur les candidats les plus prometteurs. Cependant, les essais cliniques, la phase la plus coûteuse, exigent toujours les mêmes tests rigoureux sur l'humain ; les réductions de coûts totales sont donc partielles et non transformatrices.
Quel est le gain en termes de taux de réussite grâce à l'apprentissage automatique dans la découverte de médicaments ?
Le taux de réussite de base, des essais de phase I à l'approbation, est d'environ 6,21 % pour les méthodes de développement traditionnelles. L'apprentissage automatique améliore principalement la qualité des candidats entrant dans les essais cliniques plutôt que de modifier directement les taux de réussite de ces essais. En prédisant mieux la toxicité, les effets hors cible et la pharmacocinétique avant les essais chez l'humain, l'apprentissage automatique contribue à garantir que seules les molécules les plus prometteuses accèdent aux essais de phase avancée, coûteux.
Les entreprises pharmaceutiques ont-elles besoin d'une expertise interne en apprentissage automatique ?
Les grandes entreprises pharmaceutiques constituent de plus en plus d'équipes dédiées à l'IA et au ML. Les petites entreprises de biotechnologie s'associent souvent à des sociétés spécialisées dans la découverte de médicaments par modélisation informatique ou à des groupes de recherche universitaires. L'essentiel n'est pas forcément d'embaucher des dizaines de data scientists, mais plutôt de veiller à ce que les experts en ML et les chercheurs en sciences expérimentales collaborent étroitement au lieu de travailler en vase clos.
Les modèles d'apprentissage automatique peuvent-ils remplacer entièrement les tests expérimentaux ?
Non. Les prédictions issues de l'apprentissage automatique doivent toujours être validées expérimentalement. Les modèles informatiques peuvent réduire considérablement le nombre d'expériences nécessaires en éliminant les candidats improbables, mais les tests physiques sur des cellules, des animaux et, en dernier recours, des humains, demeurent essentiels. Les agences réglementaires exigent des preuves expérimentales ; aucun médicament ne sera approuvé sur la seule base de prédictions algorithmiques.
Quels types d'algorithmes d'apprentissage automatique sont les plus performants pour la découverte de médicaments ?
Différents algorithmes excellent dans différentes tâches. Les réseaux de neurones graphiques sont performants pour la prédiction de la structure moléculaire. Les forêts aléatoires et le gradient boosting sont efficaces pour la prédiction des propriétés à partir de descripteurs moléculaires. L'apprentissage profond excelle lorsque de grands ensembles de données sont disponibles. L'apprentissage par renforcement est prometteur pour la génération de molécules de novo. La meilleure approche dépend du problème spécifique, des données disponibles et des ressources de calcul.
Comment l'apprentissage automatique gère-t-il les nouvelles cibles pathologiques avec des données limitées ?
L'apprentissage par transfert et l'apprentissage avec peu d'exemples sont utiles. Les modèles pré-entraînés sur de vastes bases de données chimiques peuvent être affinés sur de petits ensembles de données pour les maladies rares. Les graphes de connaissances intégrant diverses sources de données biologiques sont également précieux, permettant aux algorithmes d'exploiter des informations connexes même lorsque les exemples d'entraînement direct sont rares. Cependant, les cibles véritablement nouvelles, sans données analogues, demeurent un défi.
Quel est le calendrier prévu pour que les médicaments découverts grâce à l'apprentissage automatique parviennent aux patients ?
Plusieurs candidats médicaments conçus par apprentissage automatique ont fait l'objet d'essais cliniques ces dernières années, mais aucun n'a encore obtenu d'autorisation de mise sur le marché. Le délai entre la découverte et l'approbation reste de plusieurs années : l'apprentissage automatique accélère la phase de découverte, mais ne raccourcit ni la durée des essais cliniques ni celle de l'examen réglementaire. On peut s'attendre à ce que la première vague de médicaments découverts grâce à l'apprentissage automatique obtienne une autorisation de mise sur le marché entre la fin des années 2020 et le début des années 2030.
Conclusion
L'apprentissage automatique, autrefois simple curiosité expérimentale, est devenu un outil essentiel de la recherche pharmaceutique. Cette technologie répond à des problématiques bien réelles : coûts astronomiques, délais de plusieurs décennies et taux de réussite désespérément faibles qui ont entravé le développement des médicaments pendant des générations.
Les données issues de milliers de composés démontrent la capacité de l'apprentissage automatique à prédire les propriétés moléculaires, à identifier les interactions médicament-cible et à détecter les problèmes de toxicité plus tôt dans le processus de développement. Des modèles atteignant des taux de précision supérieurs à 95% dans des tâches spécifiques prouvent que la prédiction informatique a acquis une réelle utilité, et n'est plus seulement une promesse académique.
Le domaine reste confronté à des défis en matière de généralisation, d'interprétabilité et de validation. Mais la tendance est claire : les processus de découverte de médicaments intégreront de plus en plus l'apprentissage automatique, combinant l'expertise humaine à la puissance de calcul pour développer plus rapidement de meilleurs médicaments.
Pour les chercheurs, les entreprises pharmaceutiques et les patients en attente de nouveaux traitements, l'apprentissage automatique ne représente pas une solution miracle, mais un puissant accélérateur. Le travail de fond consistant à comprendre la biologie des maladies et à concevoir des thérapies efficaces demeure, mais les outils d'apprentissage automatique rendent ce travail plus efficient, plus ciblé et, en fin de compte, plus susceptible de réussir.