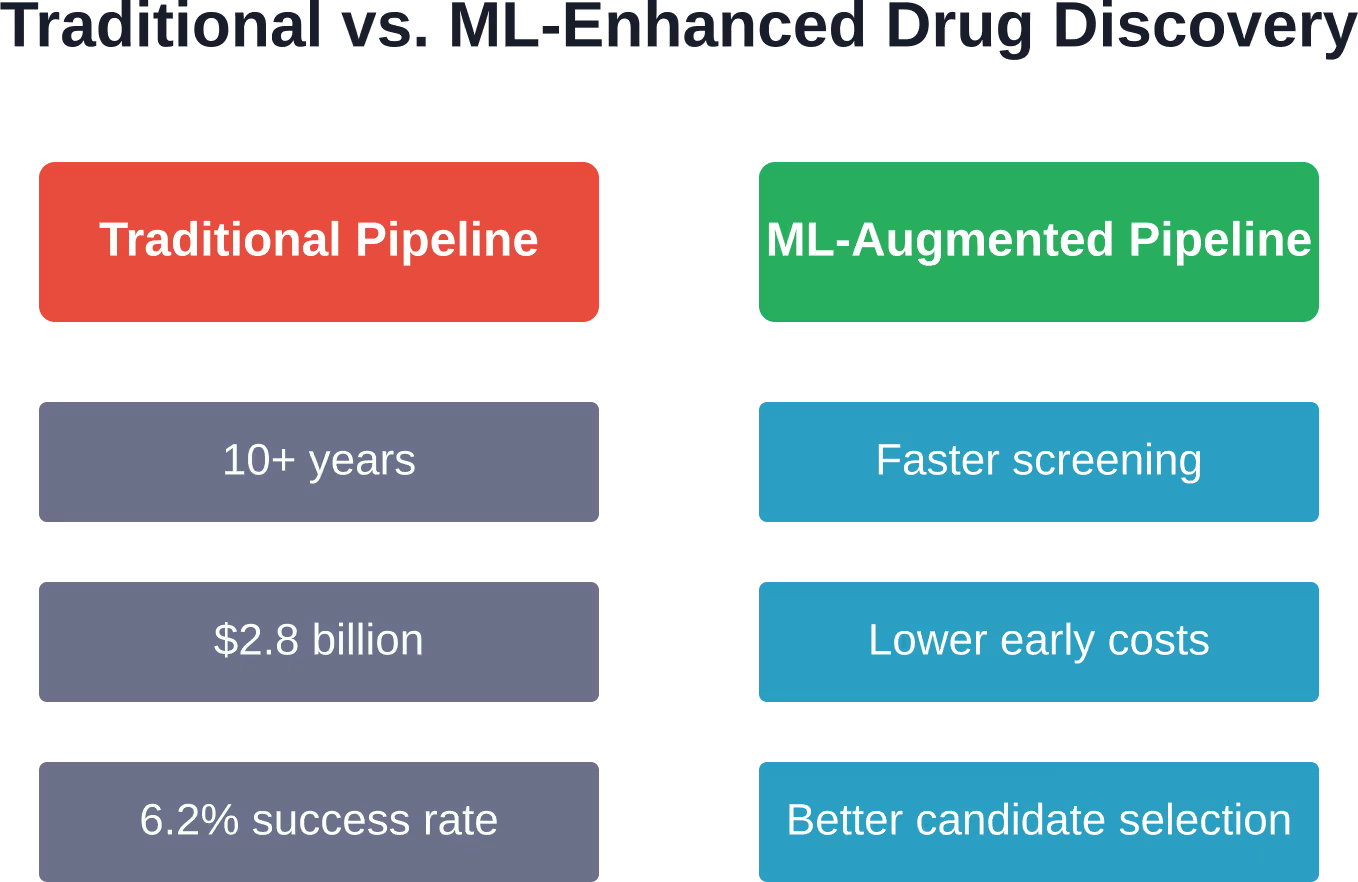
Kurzzusammenfassung: Maschinelles Lernen revolutioniert die Wirkstoffforschung, indem es das Molekül-Screening beschleunigt, Wechselwirkungen zwischen Wirkstoff und Zielstruktur vorhersagt und chemische Eigenschaften optimiert. Die Technologie begegnet der zentralen Herausforderung der Branche: Die traditionelle Arzneimittelentwicklung dauert über ein Jahrzehnt und kostet durchschnittlich 2,8 Milliarden US-Dollar, bei einer Erfolgsquote von etwa 6,21 % von Phase-I-Studien bis zur Zulassung. ML-Modelle helfen Pharmaunternehmen nun, vielversprechende Verbindungen schneller zu identifizieren, Toxizität frühzeitig vorherzusagen und kostspielige Fehlschläge in späten Entwicklungsphasen zu reduzieren.
Die Pharmaindustrie steht vor einer brutalen Realität. Die Entwicklung eines einzigen Medikaments dauert laut medizinischer Forschung über ein Jahrzehnt und kostet durchschnittlich 1,4 Milliarden US-Dollar. Selbst nach diesen enormen Investitionen scheitern neun von zehn therapeutischen Molekülen zwischen der Phase-II-Studie und der Zulassung.
Maschinelles Lernen hat sich als leistungsstarkes Werkzeug zur Bewältigung dieser gravierenden Ineffizienzen erwiesen. Durch die Analyse umfangreicher chemischer Bibliotheken, die Vorhersage des molekularen Verhaltens und die frühzeitige Identifizierung vielversprechender Wirkstoffkandidaten verändern ML-Techniken grundlegend die Herangehensweise von Forschern an die Wirkstoffentwicklung.
Die Herausforderung der Wirkstoffforschung
Die traditionelle Arzneimittelentwicklung verläuft linear und ist zeitaufwendig. Wissenschaftler screenen Tausende von Substanzen experimentell, testen sie in Zellkulturen, erproben vielversprechende Kandidaten in Tiermodellen und gehen erst dann zu klinischen Studien am Menschen über. Jede Phase erfordert jahrelange Arbeit und Millionen an Fördermitteln.
Die Zahlen sprechen eine ernüchternde Sprache. Von 21.143 untersuchten Wirkstoffen liegt die Erfolgsquote von Phase-I-Studien bis zur Zulassung bei etwa 6,21 %. Das bedeutet, dass von 100 Medikamenten, die in die klinische Prüfung am Menschen gehen, weniger als sieben in den Handel gelangen.
Wie maschinelles Lernen das Spiel verändert
Maschinelles Lernen führt einen grundlegend anderen Ansatz ein. Anstatt Verbindungen einzeln im Labor zu testen, können ML-Modelle Millionen von Molekülstrukturen rechnerisch auswerten und deren Erfolgswahrscheinlichkeit vorhersagen, bevor ein einziges Experiment durchgeführt wird.
Die Technologie zeichnet sich durch ihre Fähigkeit aus, Muster in hochdimensionalen chemischen und biologischen Daten zu erkennen – Muster, die menschlichen Forschern mit bloßem Auge verborgen bleiben. Ein neuronales Netzwerk kann die dreidimensionale Struktur eines Proteins analysieren, vorhersagen, wie Tausende kleiner Moleküle daran binden könnten, und Kandidaten nach ihrer voraussichtlichen Wirksamkeit einordnen.

Maschinelles Lernen in der Wirkstoffforschung mit überlegener KI anwenden
Maschinelles Lernen wird zur Verarbeitung großer biologischer und chemischer Datensätze und zur Unterstützung der Entscheidungsfindung in der frühen Forschungsphase eingesetzt. AI Superior bietet KI-Beratung und kundenspezifische Entwicklung von maschinellem Lernen für datengetriebene Anwendungen im Gesundheitswesen und verwandten Bereichen an.
Benötigen Sie Hilfe beim Aufbau einer ML-Lösung für die Wirkstoffforschung?
AI Superior unterstützt:
- Benutzerdefinierte Entwicklung von Modellen für maschinelles Lernen
- Datenanalyse und prädiktive Modellierung
- Lösungen für Computer Vision und Mustererkennung
- KI-Beratung und PoC-Entwicklung
👉Kontaktieren Sie AI Superior um Ihr Projekt zum maschinellen Lernen in der Wirkstoffforschung zu besprechen.
Wichtige ML-Anwendungen entlang der gesamten Pipeline
Virtuelles Screening und Hit-Entdeckung
Die früheste Phase der Wirkstoffforschung besteht darin, “Treffer” zu identifizieren – Moleküle, die eine biologische Aktivität gegen ein Krankheitsziel aufweisen. Traditionell bedeutete dies, Zehntausende von Verbindungen in Labortests zu prüfen.
ML-gestütztes virtuelles Screening kehrt dieses Modell um. Deep-Learning-Algorithmen, die mit chemischen Strukturdatenbanken trainiert wurden, können vorhersagen, welche Moleküle am wahrscheinlichsten an ein bestimmtes Protein binden. Anschließend testen die Forscher experimentell nur die vielversprechendsten Kandidaten, wodurch die Anzahl der zu synthetisierenden und zu testenden Verbindungen drastisch reduziert wird.
Vorhersage von Wechselwirkungen zwischen Wirkstoff und Zielmolekül
Das Verständnis der Wechselwirkung eines kleinen Moleküls mit seinem biologischen Zielmolekül ist für die Arzneimittelentwicklung entscheidend. Bindet es stark genug? Aktiviert oder hemmt es das Zielprotein? Verursacht es unerwünschte Nebenwirkungen?
Maschinelle Lernmodelle gehen diese Fragen mit verschiedenen Ansätzen an. Graph-Neuronale Netze können Moleküle und Proteine als mathematische Graphen darstellen und lernen, die Bindungsaffinität anhand struktureller Merkmale vorherzusagen. Rekurrente neuronale Netze mit Reinforcement Learning zeigen starke Leistungen bei Aufgaben mit Bewertungsfunktionen.
Immobilienoptimierung und Leadgenerierung
Die Suche nach einem Molekül, das ein Zielmolekül trifft, ist erst der Anfang. Dieser erste Treffer muss hinsichtlich arzneimittelähnlicher Eigenschaften optimiert werden: orale Bioverfügbarkeit, metabolische Stabilität, Blut-Hirn-Schranken-Penetration, geringe Toxizität und Herstellbarkeit.
ML-Modelle helfen dabei, sich in diesem komplexen Optimierungsfeld zurechtzufinden. Durch das Training mit Datensätzen, die chemische Strukturen mit gemessenen Eigenschaften verknüpfen, lernen Algorithmen vorherzusagen, wie sich Strukturmodifikationen auf das Verhalten einer Verbindung auswirken. Medizinische Chemiker können dann Millionen von chemischen Varianten computergestützt untersuchen, bevor sie die vielversprechendsten Optionen synthetisieren.
Klassifikationsbäume und Random-Forest-Modelle weisen in der Arzneimitteltherapieanalyse unterschiedliche Genauigkeitsgrade auf. Diese Ensemble-Methoden kombinieren mehrere Entscheidungsbäume, um auch bei fehlerhaften oder unvollständigen Trainingsdaten robuste Vorhersagen zu treffen.

Datenqualität: Die Grundlage des Erfolgs
Maschinelle Lernmodelle sind nur so gut wie die Daten, mit denen sie trainiert werden. Die Pharmaindustrie generiert seit Jahrzehnten biologische und chemische Daten, doch ein Großteil davon liegt in firmeneigenen Datenbanken oder in veröffentlichten Artikeln in Formaten vor, die eine aufwendige Bereinigung erfordern.
Die Datenaufbereitung beansprucht den größten Teil des Aufwands in jedem ML-basierten Wirkstoffforschungsprojekt. Chemische Strukturen müssen standardisiert werden. Experimentelle Messungen erfordern eine Qualitätskontrolle, um Ausreißer und Fehler zu entfernen. Biologische Testdaten müssen über verschiedene experimentelle Plattformen hinweg normalisiert werden.
Diese unscheinbare Vorverarbeitungsarbeit entscheidet über Erfolg oder Misserfolg eines Modells. Ein tiefes neuronales Netzwerk, das mit verrauschten, inkonsistenten Daten trainiert wird, liefert unzuverlässige Vorhersagen – was man hineingibt, kommt auch wieder heraus. Teams, die intensiv in die Datenaufbereitung und -validierung investieren, erzielen deutlich bessere Ergebnisse als jene, die den neuesten algorithmischen Innovationen hinterherjagen.
Aktuelle Einschränkungen und Herausforderungen
Trotz beeindruckender Fortschritte steht maschinelles Lernen in der Wirkstoffforschung vor erheblichen Herausforderungen. Modelle, die für einen bestimmten chemischen Raum trainiert wurden, versagen oft bei der Anwendung auf strukturell unterschiedliche Verbindungen. Transferlernen hilft zwar, löst das Generalisierungsproblem aber nicht vollständig.
Die Interpretierbarkeit bleibt ein zentrales Anliegen. Wenn ein neuronales Netzwerk, das nicht auf Basis von KI-Methoden funktioniert, vorhersagt, dass Verbindung X erfolgreich sein wird, Verbindung Y hingegen nicht, möchten Medizinchemiker die Gründe dafür verstehen. Zwar verbessern sich erklärbare KI-Techniken stetig, doch viele Modelle fungieren nach wie vor als undurchschaubare Orakel.
Die Branche hat auch mit der Validierung zu kämpfen. Ein Modell mag zwar auf zurückgehaltenen Testdaten eine Genauigkeit von 95% erreichen, aber lässt sich das auch auf den realen Einsatz übertragen? Die prospektive Validierung – bei der ML-Vorhersagen experimentell im Labor getestet werden – liefert den ultimativen Beweis, und viele veröffentlichte Modelle wurden dieser strengen Prüfung nicht unterzogen.
Das regulatorische Umfeld
Die FDA hat begonnen, Leitlinien zum Einsatz von KI und ML in der Arzneimittelentwicklung zu veröffentlichen. Im Januar 2025 veröffentlichte die FDA einen Leitlinienentwurf zum Einsatz künstlicher Intelligenz in der Entwicklung von Arzneimitteln und Biologika.
Diese verstärkte Aufmerksamkeit der Regulierungsbehörden birgt sowohl Chancen als auch Herausforderungen. Einerseits legitimiert die Anerkennung durch die FDA maschinelles Lernen als wertvolles Werkzeug in der pharmazeutischen Forschung. Andererseits müssen Unternehmen nun nachweisen, dass ihre KI-Systeme die Standards für Transparenz, Reproduzierbarkeit und Validierung erfüllen – Anforderungen, die die Implementierung komplexer gestalten.
| Anwendungsgebiet des maschinellen Lernens | Hauptvorteil | Hauptherausforderung |
|---|---|---|
| Virtuelles Screening | Testen Sie Millionen von Verbindungen rechnerisch | Falsch positive Vorhersagen |
| Zielvorhersage | Neue Zusammenhänge zwischen Medikamenten und Krankheiten identifizieren | Begrenzte Trainingsdaten für seltene Krankheiten |
| Immobilienoptimierung | Navigieren Sie im Designraum mit mehreren Zielen. | Ausgleich konkurrierender Eigenschaften |
| Toxizitätsvorhersage | Gefährliche Verbindungen frühzeitig kennzeichnen | Lücken im Verständnis der Toxizitätsmechanismen |
| Design klinischer Studien | Patientenstratifizierung und Endpunktauswahl | Datenschutz- und Datenweitergabebeschränkungen |
Auswirkungen in der Praxis und Fallstudien
Pharmaunternehmen und Biotech-Startups setzen maschinelles Lernen aktiv in ihren Entwicklungsprozessen ein. Große Forschungseinrichtungen bieten mittlerweile spezielle Schulungsprogramme zum maschinellen Lernen für die Wirkstoffforschung an, was die zunehmende Reife des Fachgebiets widerspiegelt.
Die akademische Forschung verschiebt kontinuierlich die Grenzen des Machbaren. Die Integration von maschinellem Lernen beschränkt sich nicht mehr auf die Grundlagenforschung. ML-Modelle unterstützen heute die Optimierung klinischer Studien, die Patientenrekrutierung, die Vorhersage von Nebenwirkungen und die Prozesskontrolle in der Arzneimittelherstellung. Der gesamte Arzneimittelentwicklungszyklus integriert zunehmend algorithmische Entscheidungshilfen.
Ausblick: Die ML-integrierte Pipeline
Die nächste Generation der Wirkstoffforschung wird maschinelles Lernen nicht als optionale Ergänzung betrachten. Vielmehr wird ML das rechnergestützte Rückgrat der pharmazeutischen Forschung bilden und in jeder Phase – von der ersten Zielidentifizierung bis zur Überwachung nach der Markteinführung – integriert sein.
Es entstehen bereits hybride Ansätze, bei denen ML-Vorhersagen die Versuchsplanung steuern und die experimentellen Ergebnisse zur Verbesserung der Modelle beitragen. Dieser iterative Zyklus beschleunigt das Lernen weit über das hinaus, was Menschen oder Algorithmen allein erreichen könnten.
Generative KI stellt die neueste Grenze dar. Anstatt lediglich bestehende Verbindungen zu screenen, können generative Modelle neuartige Molekülstrukturen entwerfen, die für spezifische Eigenschaften optimiert sind. Diese KI-designten Moleküle erschließen oft chemische Bereiche, die menschliche Chemiker intuitiv nicht in Betracht ziehen würden, und führen so zu wahrhaft innovativen Therapeutika.
Technologie allein wird die Herausforderungen der Wirkstoffforschung nicht lösen. Erfolg erfordert die Zusammenarbeit von Datenwissenschaftlern mit Kenntnissen im Bereich maschinelles Lernen und Fachexperten aus Biologie, Chemie und Medizin. Die effektivsten Teams kombinieren Rechenleistung mit fundiertem wissenschaftlichem Wissen.
Häufig gestellte Fragen
Um wie viel reduziert maschinelles Lernen die Kosten der Arzneimittelentwicklung?
Maschinelles Lernen (ML) kann die Kosten in der frühen Entwicklungsphase deutlich senken, indem es die Anzahl der Verbindungen reduziert, die physikalisch synthetisiert und getestet werden müssen. Während die traditionelle Pipeline durchschnittlich 1,4 Milliarden US-Dollar pro zugelassenem Medikament kostet, ermöglicht das ML-gestützte Screening Forschern, ihre experimentellen Ressourcen auf die vielversprechendsten Kandidaten zu konzentrieren. Klinische Studien – die teuerste Phase – erfordern jedoch weiterhin die gleichen strengen Tests am Menschen, sodass die Gesamtkostensenkungen eher partiell als grundlegend sind.
Welche Verbesserung der Erfolgsquote ergibt sich durch den Einsatz von ML in der Wirkstoffforschung?
Die durchschnittliche Erfolgsrate von Phase-I-Studien bis zur Zulassung liegt bei der traditionellen Entwicklung bei etwa 6,21 %. Maschinelles Lernen (ML) verbessert primär die Qualität der Kandidaten für klinische Studien, anstatt die Erfolgsraten direkt zu beeinflussen. Durch die bessere Vorhersage von Toxizität, Off-Target-Effekten und Pharmakokinetik vor klinischen Studien trägt ML dazu bei, dass nur die vielversprechendsten Moleküle in die kostenintensiven späten Phasen gelangen.
Benötigen Pharmaunternehmen interne Expertise im Bereich maschinelles Lernen?
Große Pharmaunternehmen bauen zunehmend eigene KI- und ML-Teams auf. Kleinere Biotech-Firmen kooperieren häufig mit spezialisierten Unternehmen für computergestützte Wirkstoffforschung oder akademischen Forschungsgruppen. Entscheidend ist nicht unbedingt die Einstellung Dutzender Data Scientists, sondern die enge Zusammenarbeit von ML-Experten und experimentellen Wissenschaftlern, anstatt isoliert voneinander zu arbeiten.
Können ML-Modelle experimentelle Tests vollständig ersetzen?
Nein. Vorhersagen des maschinellen Lernens müssen stets experimentell validiert werden. Computermodelle können die Anzahl der benötigten Experimente durch das Aussortieren unwahrscheinlicher Kandidaten drastisch reduzieren, doch physikalische Tests an Zellen, Tieren und letztendlich Menschen bleiben unerlässlich. Zulassungsbehörden fordern experimentelle Nachweise; kein Medikament wird allein auf Basis algorithmischer Vorhersagen zugelassen.
Welche Arten von ML-Algorithmen eignen sich am besten für die Wirkstoffforschung?
Verschiedene Algorithmen eignen sich hervorragend für unterschiedliche Aufgaben. Graph-Neuronale Netze eignen sich gut für die Vorhersage von Molekülstrukturen. Random Forests und Gradient Boosting sind effektiv für die Vorhersage von Eigenschaften anhand von Moleküldeskriptoren. Deep Learning glänzt bei großen Datensätzen. Reinforcement Learning zeigt vielversprechende Ansätze für die Neugenerierung von Molekülen. Der beste Ansatz hängt vom jeweiligen Problem, den verfügbaren Daten und den Rechenressourcen ab.
Wie geht maschinelles Lernen mit neuartigen Krankheitszielen bei begrenzten Daten um?
Transferlernen und Few-Shot-Learning-Verfahren sind hilfreich. Modelle, die auf großen chemischen Datenbanken vortrainiert wurden, können anhand kleiner Datensätze für seltene Krankheiten feinabgestimmt werden. Wissensgraphen, die verschiedene biologische Datenquellen integrieren, sind ebenfalls nützlich, da sie es Algorithmen ermöglichen, verwandte Informationen zu nutzen, selbst wenn nur wenige direkte Trainingsbeispiele vorliegen. Dennoch bleiben wirklich neuartige Zielstrukturen ohne vergleichbare Daten eine Herausforderung.
Wie lange dauert es, bis mithilfe von maschinellem Lernen entdeckte Medikamente Patienten erreichen?
Mehrere mithilfe von maschinellem Lernen entwickelte Wirkstoffkandidaten haben in den letzten Jahren klinische Studien durchlaufen, jedoch hat noch keiner die Zulassung erhalten. Der Zeitraum von der Entdeckung bis zur Zulassung erstreckt sich weiterhin über Jahre – maschinelles Lernen beschleunigt zwar die Entdeckungsphase, verkürzt aber weder die Dauer klinischer Studien noch die behördliche Prüfung. Es ist zu erwarten, dass die ersten mithilfe von maschinellem Lernen entdeckten Medikamente Ende der 2020er und Anfang der 2030er Jahre zugelassen werden.
Schlussfolgerung
Maschinelles Lernen hat sich von einer experimentellen Kuriosität zu einem unverzichtbaren Werkzeug in der pharmazeutischen Forschung entwickelt. Die Technologie adressiert reale Probleme: astronomische Kosten, jahrzehntelange Entwicklungszeiten und deprimierend niedrige Erfolgsquoten, die die Arzneimittelentwicklung seit Generationen plagen.
Daten von Tausenden von Verbindungen belegen die Fähigkeit des maschinellen Lernens, molekulare Eigenschaften vorherzusagen, Wechselwirkungen zwischen Wirkstoff und Zielstruktur zu identifizieren und Toxizitätsprobleme frühzeitig im Entwicklungsprozess zu erkennen. Modelle mit Genauigkeitsraten von über 95% in spezifischen Aufgaben zeigen, dass die computergestützte Vorhersage nicht nur ein akademisches Versprechen ist, sondern auch einen echten praktischen Nutzen erlangt hat.
Das Feld steht weiterhin vor Herausforderungen hinsichtlich Generalisierung, Interpretierbarkeit und Validierung. Die Entwicklung ist jedoch eindeutig: Die Wirkstoffforschung wird maschinelles Lernen zunehmend integrieren und menschliches Fachwissen mit Rechenleistung kombinieren, um bessere Medikamente schneller zu entwickeln.
Für Forscher, Pharmaunternehmen und Patienten, die auf neue Therapien warten, ist maschinelles Lernen zwar keine Wunderlösung, aber ein wirkungsvoller Beschleuniger. Die anspruchsvolle Aufgabe, die Krankheitsbiologie zu verstehen und wirksame Therapien zu entwickeln, bleibt bestehen, doch ML-Werkzeuge machen diese Arbeit effizienter, zielgerichteter und erhöhen letztendlich die Erfolgswahrscheinlichkeit.