Quick Summary: Machine learning is revolutionizing drug discovery by accelerating molecule screening, predicting drug-target interactions, and optimizing chemical properties. The technology addresses the industry’s core challenge: traditional drug development takes over a decade and costs US$2.8 billion on average, with approximately 6.2% success rate from Phase I trials to approval. ML models now help pharmaceutical companies identify promising compounds faster, predict toxicity earlier, and reduce costly late-stage failures.
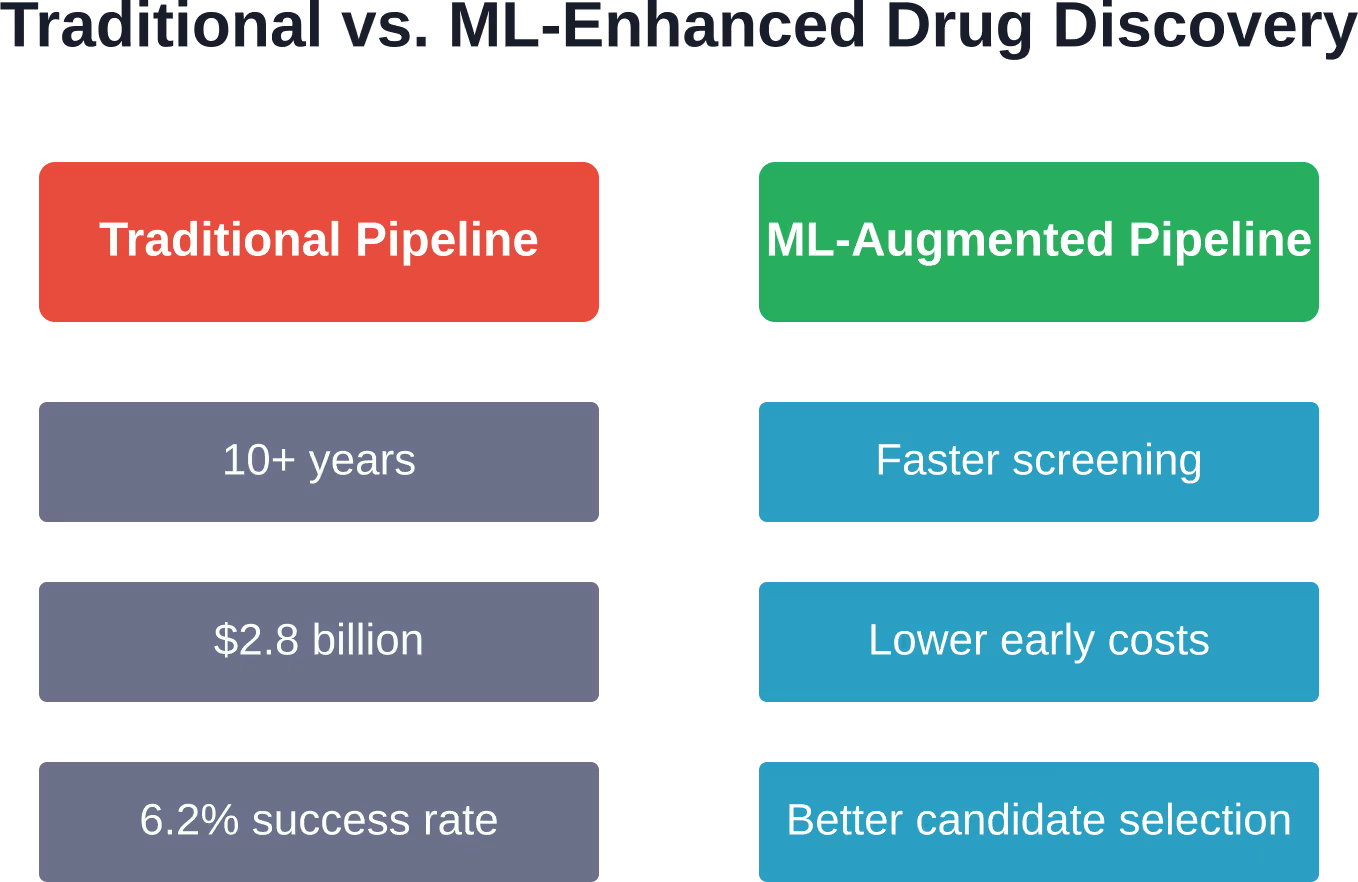
The pharmaceutical industry faces a brutal reality. Developing a single drug takes over a decade and costs US$2.8 billion on average, according to medical research. Even after that massive investment, 9 out of 10 therapeutic molecules fail between Phase II clinical trials and regulatory approval.
Machine learning has emerged as a powerful tool to tackle these staggering inefficiencies. By analyzing vast chemical libraries, predicting molecular behavior, and identifying promising drug candidates earlier in the pipeline, ML techniques are fundamentally changing how researchers approach drug discovery.
The Drug Discovery Challenge
Traditional drug development follows a linear, time-intensive process. Scientists screen thousands of compounds experimentally, test them in cell cultures, move promising candidates to animal models, and only then advance to human trials. Each stage requires years of work and millions in funding.
The numbers tell a sobering story. Across 21,143 compounds studied, the overall success rate from Phase I clinical trials to drug approval is approximately 6.2%. That means for every 100 drugs that enter human testing, fewer than seven make it to pharmacy shelves.
How Machine Learning Changes the Game
Machine learning introduces a fundamentally different approach. Instead of testing compounds one by one in the lab, ML models can evaluate millions of molecular structures computationally, predicting their likelihood of success before a single experiment runs.
The technology excels at finding patterns in high-dimensional chemical and biological data—patterns that human researchers simply can’t spot by eye. A neural network can analyze the three-dimensional structure of a protein, predict how thousands of small molecules might bind to it, and rank candidates by their predicted effectiveness.

Apply Machine Learning to Drug Discovery With AI Superior
Machine learning is used to process large biological and chemical datasets and support decision-making in early-stage research. AI Superior provides AI consulting and custom machine learning development for data-driven applications in healthcare and related domains.
Need Help Building a Drug Discovery ML Solution?
AI Superior supports:
- Custom machine learning model development
- Data analysis and predictive modeling
- Computer vision and pattern recognition solutions
- AI consulting and PoC development
👉Contact AI Superior to discuss your drug discovery machine learning project.
Key ML Applications Across the Pipeline
Virtual Screening and Hit Discovery
The earliest stage of drug discovery involves identifying “hits”—molecules that show any biological activity against a disease target. Traditionally, this meant physically testing tens of thousands of compounds in laboratory assays.
ML-powered virtual screening flips this model. Deep learning algorithms trained on chemical structure databases can predict which molecules are most likely to bind to a specific protein target. Researchers then test only the top-ranked candidates experimentally, dramatically reducing the number of compounds that need synthesis and testing.
Drug-Target Interaction Prediction
Understanding how a small molecule interacts with its biological target is critical for drug development. Does it bind strongly enough? Does it activate or inhibit the target protein? Will it cause off-target effects?
Machine learning models tackle these questions through multiple approaches. Graph neural networks can represent molecules and proteins as mathematical graphs, learning to predict binding affinity from structural features. Recurrent neural networks with reinforcement learning show strong performance in scoring function tasks.
Property Optimization and Lead Development
Finding a molecule that hits a target is just the beginning. That initial “hit” must be optimized for drug-like properties: oral bioavailability, metabolic stability, blood-brain barrier penetration, low toxicity, and manufacturability.
ML models help navigate this complex optimization landscape. By training on datasets linking chemical structures to measured properties, algorithms learn to predict how structural modifications will affect a compound’s behavior. Medicinal chemists can then explore millions of chemical variants computationally before synthesizing the most promising options.
Classification trees and random forest models show varying accuracy levels in drug treatment analysis. These ensemble methods combine multiple decision trees to produce robust predictions even when training data is noisy or incomplete.

Data Quality: The Foundation of Success
Machine learning models are only as good as the data they’re trained on. The pharmaceutical industry has spent decades generating biological and chemical data, but much of it sits in proprietary databases or published papers in formats that require extensive cleaning.
Data preparation consumes the majority of effort in any ML drug discovery project. Chemical structures need standardization. Experimental measurements require quality control to remove outliers and errors. Biological assay data must be normalized across different experimental platforms.
This unglamorous preprocessing work determines whether a model succeeds or fails. A deep neural network trained on noisy, inconsistent data will produce unreliable predictions—garbage in, garbage out. Teams that invest heavily in data curation and validation consistently outperform those chasing the latest algorithmic innovations.
Current Limitations and Challenges
Despite impressive advances, ML in drug discovery faces significant hurdles. Models trained on one type of chemical space often fail when applied to structurally different compounds. Transfer learning helps but doesn’t fully solve the generalization problem.
Interpretability remains a major concern. When a black-box neural network predicts that compound X will succeed while compound Y will fail, medicinal chemists want to understand why. Explainable AI techniques are improving, but many models still function as inscrutable oracles.
The industry also struggles with validation. A model might achieve 95% accuracy on held-out test data, but does that translate to real-world success? Prospective validation—where ML predictions are tested experimentally in the lab—provides the ultimate proof, and many published models haven’t undergone this rigorous scrutiny.
The Regulatory Landscape
The FDA has begun issuing guidance on AI and ML use in drug development. In January 2025, the FDA released draft guidance on the use of artificial intelligence for drug and biological product development.
This regulatory attention signals both opportunity and challenge. On one hand, FDA acknowledgment legitimizes ML as a valuable tool in pharmaceutical research. On the other, companies must now demonstrate that their AI systems meet standards for transparency, reproducibility, and validation—requirements that add complexity to deployment.
| ML Application Area | Primary Benefit | Key Challenge |
|---|---|---|
| Virtual Screening | Test millions of compounds computationally | False positive predictions |
| Target Prediction | Identify novel drug-disease connections | Limited training data for rare diseases |
| Property Optimization | Navigate multi-objective design space | Balancing competing properties |
| Toxicity Prediction | Flag dangerous compounds early | Gaps in toxicity mechanism understanding |
| Clinical Trial Design | Patient stratification and endpoint selection | Privacy and data sharing constraints |
Real-World Impact and Case Studies
Pharmaceutical companies and biotech startups are actively deploying ML across their pipelines. Major research institutions now offer dedicated training programs on machine learning for drug discovery, reflecting the field’s maturation.
Academic research continues to push boundaries. The integration of machine learning isn’t limited to early-stage research. ML models now assist with clinical trial optimization, patient recruitment, adverse event prediction, and manufacturing process control. The entire drug development lifecycle increasingly incorporates algorithmic decision support.
Looking Forward: The ML-Integrated Pipeline
The next generation of drug discovery won’t treat machine learning as an optional add-on. Instead, ML will form the computational backbone of pharmaceutical research, integrated at every stage from initial target identification through post-market surveillance.
Hybrid approaches are already emerging where ML predictions guide experimental design, and experimental results feed back to improve models. This iterative cycle accelerates learning far beyond what either humans or algorithms could achieve alone.
Generative AI represents the newest frontier. Rather than merely screening existing compounds, generative models can design novel molecular structures optimized for specific properties. These AI-designed molecules often explore chemical space that human chemists wouldn’t intuitively consider, leading to genuinely innovative therapeutics.
But technology alone won’t solve drug discovery’s challenges. Success requires collaboration between data scientists who understand ML and domain experts who understand biology, chemistry, and medicine. The most effective teams combine computational power with deep scientific knowledge.
Frequently Asked Questions
How much does machine learning reduce drug development costs?
ML can significantly lower early-stage costs by reducing the number of compounds that need physical synthesis and testing. While the traditional pipeline costs US$2.8 billion on average per approved drug, ML-enhanced screening lets researchers focus experimental resources on the most promising candidates. However, clinical trials—the most expensive phase—still require the same rigorous human testing, so total cost reductions are partial rather than transformative.
What’s the success rate improvement with ML in drug discovery?
The baseline success rate from Phase I trials to approval is approximately 6.2% across traditional development. ML primarily improves the quality of candidates entering clinical trials rather than changing trial success rates directly. By better predicting toxicity, off-target effects, and pharmacokinetics before human testing, ML helps ensure that only the most promising molecules advance to expensive late-stage trials.
Do pharmaceutical companies need in-house ML expertise?
Large pharmaceutical companies increasingly build dedicated AI and ML teams. Smaller biotech firms often partner with specialized computational drug discovery companies or academic research groups. The key isn’t necessarily hiring dozens of data scientists—it’s ensuring that ML experts and experimental scientists collaborate closely rather than working in silos.
Can ML models replace experimental testing entirely?
No. ML predictions must always be validated experimentally. Computational models can dramatically reduce the number of experiments needed by filtering out unlikely candidates, but physical testing in cells, animals, and ultimately humans remains essential. Regulatory agencies require experimental evidence; no drug will be approved based solely on algorithmic predictions.
What types of ML algorithms work best for drug discovery?
Different algorithms excel at different tasks. Graph neural networks handle molecular structure prediction well. Random forests and gradient boosting work effectively for property prediction from molecular descriptors. Deep learning shines when large datasets are available. Reinforcement learning shows promise for de novo molecule generation. The best approach depends on the specific problem, available data, and computational resources.
How does ML handle novel disease targets with limited data?
Transfer learning and few-shot learning techniques help. Models pre-trained on large chemical databases can be fine-tuned on small datasets for rare diseases. Knowledge graphs that integrate diverse biological data sources also help, letting algorithms leverage related information even when direct training examples are scarce. Still, truly novel targets with no analogous data remain challenging.
What’s the timeline for ML-discovered drugs to reach patients?
Several ML-designed drug candidates have entered clinical trials in recent years, but none have yet achieved regulatory approval. The timeline from discovery to approval still spans years—ML accelerates the discovery phase but doesn’t shorten clinical trial durations or regulatory review. Expect to see the first wave of ML-discovered drugs reaching approval in the late 2020s and early 2030s.
Conclusion
Machine learning has moved from experimental curiosity to an essential tool in pharmaceutical research. The technology addresses real pain points: astronomical costs, decade-long timelines, and depressingly low success rates that have plagued drug development for generations.
Data from thousands of compounds demonstrates ML’s ability to predict molecular properties, identify drug-target interactions, and flag toxicity issues earlier in the pipeline. Models achieving accuracy rates above 95% in specific tasks show that computational prediction has reached genuine utility, not just academic promise.
The field still faces challenges around generalization, interpretability, and validation. But the trajectory is clear—drug discovery pipelines will continue integrating ML more deeply, combining human expertise with computational power to develop better medicines faster.
For researchers, pharmaceutical companies, and patients waiting for new treatments, machine learning represents not a magic solution but a powerful accelerant. The hard work of understanding disease biology and designing effective therapies remains, but ML tools make that work more efficient, more targeted, and ultimately more likely to succeed.