Quick Summary: Machine learning is transforming materials science by accelerating discovery, predicting properties, and optimizing designs that once took years to develop. Researchers now train algorithms on vast materials databases to predict formation energies, recommend synthesis routes, and classify microstructures with accuracies exceeding 98%. This computational revolution—backed by government initiatives including a $100 million NSF, in partnership with Capital One and Intel investment in AI research institutes—enables scientists to screen thousands of candidate materials in hours, fundamentally changing how we develop everything from batteries to structural alloys.
Materials science has always been a waiting game. Discovering a new alloy, ceramic, or polymer traditionally meant years of trial-and-error experiments, expensive fabrication runs, and painstaking characterization. But that timeline is collapsing.
Machine learning algorithms now analyze decades of research data in seconds, predict material properties before synthesis, and recommend promising candidates from millions of possibilities. According to the National Institute of Standards and Technology (NIST), these AI-driven workflows are demonstrating accelerated material development from discovery through commercialization and even circularity.
The shift is not subtle. Where human intuition recommended successful synthesis routes for inorganic-organic hybrid materials 78% of the time, a support vector machine (SVM) model achieved 89% accuracy. That eleven-point jump represents countless saved experiments and faster routes to market.
The Foundation: Why Machine Learning Works for Materials
Materials science generates enormous datasets. Every experiment produces measurements—diffraction patterns, spectroscopy results, mechanical properties, thermal behavior. Researchers have accumulated millions of these observations across decades.
Machine learning thrives on exactly this situation: large volumes of structured data with complex, non-linear relationships. The atomic composition of a material determines its crystal structure, which influences electronic properties, which affect mechanical behavior. These connections are too intricate for simple equations but perfect for neural networks.
Here’s the thing though—materials data is uniquely well-suited to ML because the underlying physics constrains the solution space. Unlike predicting stock prices or social trends, materials obey conservation laws, thermodynamic principles, and quantum mechanics. Machine learning models learn these patterns implicitly from data.
The National Science Foundation has invested in artificial intelligence research since the early 1960s, setting technical foundations that drive today’s innovations. On July 29, 2025, NSF, in partnership with Capital One and Intel, announced a $100 million investment to support five National Artificial Intelligence Research Institutes, including the NSF AI-Materials Institute (NSF AI-MI) led by Cornell University.
The Data Advantage in Materials Research
Materials databases have grown exponentially. The Materials Project, AFLOW, and NIST’s own repositories contain calculated and experimental data for hundreds of thousands of compounds. This scale enables training sophisticated models.
Consider formation enthalpy—the energy released or absorbed when a compound forms from its elements. A deep neural network called ElemNet, trained on approximately 2 × 10⁵ compounds, achieved a mean absolute error of just 0.050 ± 0.0007 eV/atom when tested on roughly 2 × 10⁴ different compounds. This performance allows researchers to screen candidates rapidly.
The architecture matters. ElemNet uses 17 layers to extract progressively abstract features from raw elemental compositions. Early layers might recognize electronegativity differences, while deeper layers capture complex bonding tendencies. This hierarchical learning mirrors how materials scientists think about structure-property relationships.
Key Applications Transforming Materials Development
Machine learning has moved from academic curiosity to production tool across multiple materials domains. The applications fall into several categories, each with measurable impact.
Property Prediction Before Synthesis
The most immediate value proposition: predict whether a material will have desired properties before spending time and money making it. Neural networks trained on density functional theory (DFT) calculations can estimate band gaps, elastic moduli, thermal conductivity, and dozens of other properties from chemical formula alone.
This capability compresses the discovery cycle. Instead of synthesizing 100 candidates to find one with the right combination of strength and conductivity, researchers screen 10,000 computationally, synthesize the top 10, and find three winners.
Real talk: this doesn’t eliminate experiments. ML predictions have uncertainty, and real materials have defects, grain boundaries, and processing history that pure composition can’t capture. But it shifts the experimental bottleneck from broad exploration to targeted validation.
| Property Type | ML Approach | Typical Accuracy | Training Data Size |
|---|---|---|---|
| Formation Energy | Deep Neural Networks | ~9% MAE | 100k–200k compounds |
| Band Gap | Graph Neural Networks | 0.2–0.4 eV MAE | 50k–100k compounds |
| Elastic Modulus | Random Forests | 10–15% error | 10k–30k samples |
| Thermal Conductivity | Gradient Boosting | 15–25% error | 5k–15k samples |
Microstructure Classification and Analysis
Materials scientists spend significant time examining micrographs—images from optical, electron, or atomic force microscopes that reveal grain structures, phases, and defects. Classifying these images traditionally required expert judgment and was inherently subjective.
Convolutional neural networks (CNNs) automate this process with remarkable fidelity. Transfer learning—taking a network pre-trained on millions of everyday images and fine-tuning it on materials micrographs—achieves impressive results even with limited training data.
Transfer learning with CNN architectures has achieved microstructure classification accuracies exceeding 98% in published research. These aren’t toy problems; accurate microstructure classification connects processing conditions to performance, enabling better quality control and process optimization.
The implications extend beyond classification. Once a network learns what different phases look like, it can quantify their distributions, track evolution during heat treatment, and correlate microstructural features with mechanical properties. This closes the loop between processing, structure, and performance.
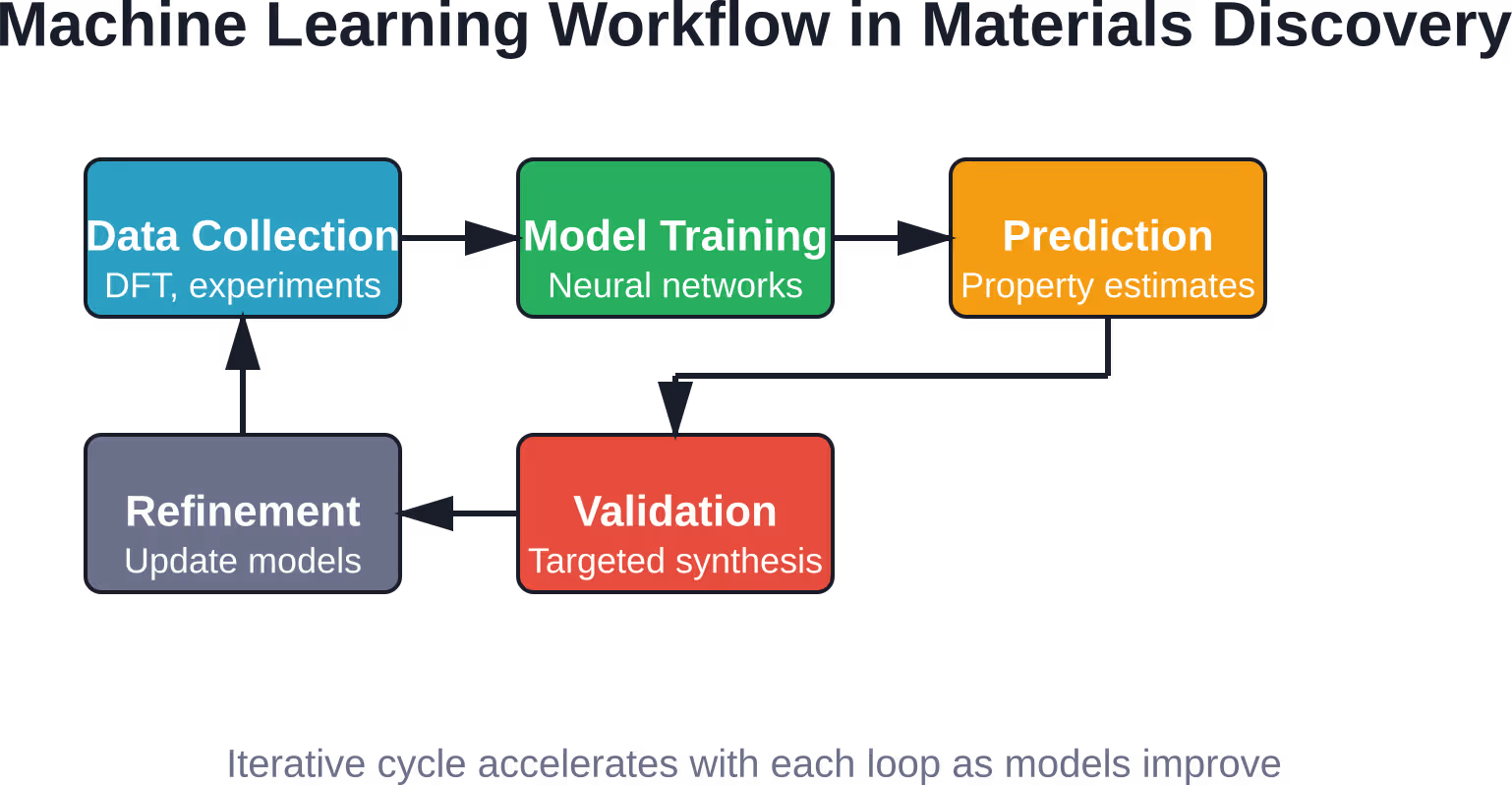
Synthesis Route Recommendation
Knowing what material you want is one thing. Figuring out how to make it is another. Synthesis involves choosing precursors, solvents, temperatures, reaction times, and atmospheres. The combinatorial explosion of possibilities is staggering.
Machine learning models trained on experimental notes and synthesis reports can recommend likely successful routes. For inorganic-organic hybrid materials, researchers built an SVM model that achieved an 89% reaction recommendation success rate compared to 78% for human intuition—a significant improvement that translates to fewer failed batches and faster optimization.
These models learn from both successes and failures. A reaction that didn’t produce the target phase still provides information about what conditions to avoid. Natural language processing techniques extract reaction parameters from published literature, building training datasets automatically.
Accelerated Characterization with Virtual Spectroscopy
Characterizing materials requires expensive instruments—X-ray diffractometers, infrared spectrometers, electron microscopes. Each modality provides different information, and comprehensive characterization needs multiple techniques.
MIT researchers developed SpectroGen, an AI tool that functions as a virtual spectrometer. Feed it a spectrum from one modality (say, infrared), and it generates what that material’s spectrum would look like in a different modality (like X-ray). The AI-generated results match physical measurements with 99% accuracy and complete predictions in less than one minute.
This capability dramatically reduces characterization costs and time. A manufacturer can perform one quick measurement and use SpectroGen to generate predicted spectra across multiple modalities, flagging quality issues or unexpected phases without needing access to every instrument. For industries producing materials at scale, this represents enormous efficiency gains.

Use Machine Learning in Materials Science With AI Superior
Materials science research often produces large datasets from simulations, testing environments, and laboratory experiments. AI Superior can help teams structure machine learning projects for material analysis, predictive modeling, and research automation. Their work includes AI consulting, data science, machine learning engineering, proof of concept development, and AI software support.
AI Superior can help materials science projects through:
- Preparation and evaluation of materials datasets
- Development of ML models for material analysis
- Building predictive workflows for research environments
- Detection of irregularities and material behavior patterns
- Validation and testing of analytical models
- Planning integration into internal research systems
For materials science, this may include defect analysis, material property prediction, simulation support, and experimental data processing.
👉Contact AI Superior to explore the project setup and next steps.
Deep Learning Architectures for Materials Problems
Not all machine learning models are created equal. Materials applications have gravitated toward specific architectures that handle the unique characteristics of materials data.
Graph Neural Networks for Crystal Structures
Crystals are inherently graph-like: atoms are nodes, bonds are edges, and the network topology encodes structure. Graph neural networks (GNNs) operate directly on this representation, making them natural for crystalline materials.
A GNN processes a crystal by iteratively updating each atom’s representation based on its neighbors. After several message-passing rounds, the network builds up a representation that captures local bonding environments, medium-range structural motifs, and global symmetry—all relevant to properties.
GNNs have shown particular strength predicting properties tied to electronic structure and bonding. They outperform traditional descriptor-based models for band gaps, formation energies, and magnetic properties because they directly encode geometric and chemical relationships rather than relying on hand-crafted features.
Transfer Learning and Pre-Training Strategies
Materials datasets, while large by scientific standards, are small compared to the millions of images or billions of text tokens used to train general-purpose AI. Transfer learning addresses this gap.
A network pre-trained on a large, general dataset learns broadly useful features—edge detection for images, or general chemical relationships for molecules. Fine-tuning this network on a smaller, specialized materials dataset adapts those features to the target task.
Self-supervised pre-training offers another route. A model learns by predicting masked properties or reconstructing corrupted structures. Research published in Nature demonstrated that self-supervised pre-training improved performance on material property prediction by 6.67% in terms of Mean Absolute Error (MAE).
Multi-Task and Multi-Fidelity Learning
Materials properties are correlated. A material with high melting point often has high hardness. Thermal and electrical conductivity frequently track together in metals. Multi-task learning exploits these correlations by training a single model to predict multiple properties simultaneously.
The shared representation learned across tasks captures underlying chemical and structural factors that influence all properties. This approach often outperforms separate single-task models, especially when training data for some properties is sparse—the model borrows statistical strength from related tasks.
Multi-fidelity learning addresses another material challenge: mixing data from different sources. High-fidelity DFT calculations are accurate but expensive; empirical models are fast but approximate. A multi-fidelity model learns to use cheap, low-fidelity data to correct biases and supplement expensive high-fidelity data, maximizing information extraction from available resources.
The Explainability Challenge and XAI Solutions
Here’s the problem: the most accurate machine learning models are often black boxes. A deep neural network with millions of parameters can predict material properties brilliantly but offers no insight into why. For researchers trying to understand physical principles, this is frustrating.
Explainable artificial intelligence (XAI) tackles this issue. The goal isn’t just accurate predictions but interpretable ones that reveal chemical insights and build scientific understanding.
What “Explain” Means in Materials Context
Explanation takes different forms depending on the audience and application. For a synthesis researcher, explanation might mean identifying which reaction parameters most influence yield. For a theorist, it might mean revealing which electronic structure features determine stability.
Feature importance methods rank input variables by their contribution to predictions. SHAP (SHapley Additive exPlanations) values, derived from game theory, provide a principled way to assign credit to each input feature for each prediction. If a model predicts high conductivity, SHAP reveals which elements and structural features drove that prediction.
Attention mechanisms in neural networks offer another route to interpretability. The model explicitly learns which parts of the input (which atoms, which bonds) are relevant for each property. Visualizing these attention weights highlights structural motifs that control behavior.
Balancing Accuracy and Interpretability
A tension exists between accuracy and interpretability. Linear models are transparent but often inaccurate. Deep networks are accurate but opaque. The practical solution usually involves compromise.
One strategy: use a complex model for predictions but fit a simpler, interpretable model to approximate its behavior locally. LIME (Local Interpretable Model-agnostic Explanations) implements this idea, building local linear approximations around individual predictions. The approximation explains that specific prediction, even if the underlying model is complex.
Another approach builds interpretability into the architecture. Neural networks with specialized layers that encode known physics—conservation laws, symmetry constraints, domain-specific descriptors—are both more accurate and more interpretable than generic architectures because their structure reflects real material science concepts.
| XAI Method | Interpretation Type | Best For | Limitation |
|---|---|---|---|
| SHAP Values | Feature importance | Understanding drivers | Computationally expensive |
| Attention Visualization | Structural motifs | Identifying key features | Architecture-specific |
| LIME | Local linear approximation | Individual predictions | Only local validity |
| Saliency Maps | Input sensitivity | Image/structure data | Can be noisy |
Government and Institutional Initiatives Driving Progress
Machine learning in materials science isn’t just academic research—it’s a strategic priority for governments and major institutions recognizing its economic and security implications.
NIST’s Data and AI-Driven Materials Science Efforts
The National Institute of Standards and Technology has established a dedicated Data and AI-Driven Materials Science Group developing methods, algorithms, data, and tools to accelerate discovery, development, commercialization, and circularity of industrially-relevant materials.
NIST runs an annual Machine Learning for Materials Research Bootcamp offering four days of lectures and hands-on exercises. Topics range from Python basics and data preprocessing through advanced ML techniques, providing practical training that equips researchers to apply these methods.
Setting standards is critical. NIST researchers recently published guidance on standards for data-driven materials science, addressing data quality, model validation, and reproducibility—foundational issues that must be solved for ML to deliver reliable industrial applications.
NSF’s National AI Research Infrastructure
The National Science Foundation leads the National Artificial Intelligence Research Resource (NAIRR), a national infrastructure providing research and education communities with access to computing, software, data, models, and expertise needed for AI innovation.
NAIRR focuses on expanding access to AI resources across the research community, including materials scientists. The NSF’s NAIRR Classroom component develops AI-ready workforce capabilities through education, training, and outreach to new and nontraditional communities.
This infrastructure approach recognizes that cutting-edge ML research requires computational resources beyond what individual universities typically provide. Democratizing access ensures materials innovation isn’t limited to a handful of well-funded institutions.
International Collaboration and Competition
Materials science is inherently international. The Materials Genome Initiative in the United States parallels similar efforts in Europe, Japan, and China. Machine learning has become a competitive advantage in this landscape.
Countries building superior materials databases, training more AI-capable materials scientists, and developing better computational infrastructure gain advantages in industries from aerospace to electronics to energy. The $100 million investment announced on July 29, 2025 explicitly aims to strengthen U.S. global competitiveness and accelerate innovation.
Real-World Applications and Case Studies
Enough theory. What are organizations actually doing with these techniques?
Battery Materials Optimization
Battery companies need to understand how manufacturing parameters affect cell performance. Machine learning models map these relationships, enabling optimization for specific applications, cost reduction, and yield improvement.
Researchers at Stanford and elsewhere apply ML to characterize and design battery electrodes, analyzing how composition, particle size distribution, porosity, and binder content influence capacity, rate capability, and cycle life. These models accelerate the iterative design process that traditionally required hundreds of experimental batches.
Autonomous Materials Discovery Platforms
MIT’s CRESt (Closed-Loop Robotic Experimental Search Technology) platform represents the next evolution: fully autonomous discovery. The system combines ML with robotic synthesis and characterization to run experiments, analyze results, update models, and design the next experiments—all without human intervention.
CRESt learns from diverse scientific information—literature, databases, experimental results—and generates solutions to energy problems that have plagued materials science for decades. This closed-loop approach discovers materials orders of magnitude faster than human-guided research.
The system doesn’t just predict. It actively explores, balancing exploitation (synthesizing materials it predicts will work) with exploration (testing uncertain candidates that might reveal new knowledge). This strategy, borrowed from reinforcement learning, efficiently navigates vast search spaces.
Quality Control in Manufacturing
Industrial materials production requires consistent quality. Machine learning models monitor real-time sensor data during manufacturing, predicting properties and flagging deviations before they become costly failures.
One application predicts the hardness of low-alloy metals from composition and processing parameters. Instead of waiting for post-production testing, the model provides instant feedback, enabling process adjustments that keep production within specifications.
SpectroGen’s virtual spectroscopy finds natural application here. A single quick measurement followed by AI-generated multi-modal spectra provides comprehensive quality assessment in under one minute—fast enough for production line integration.
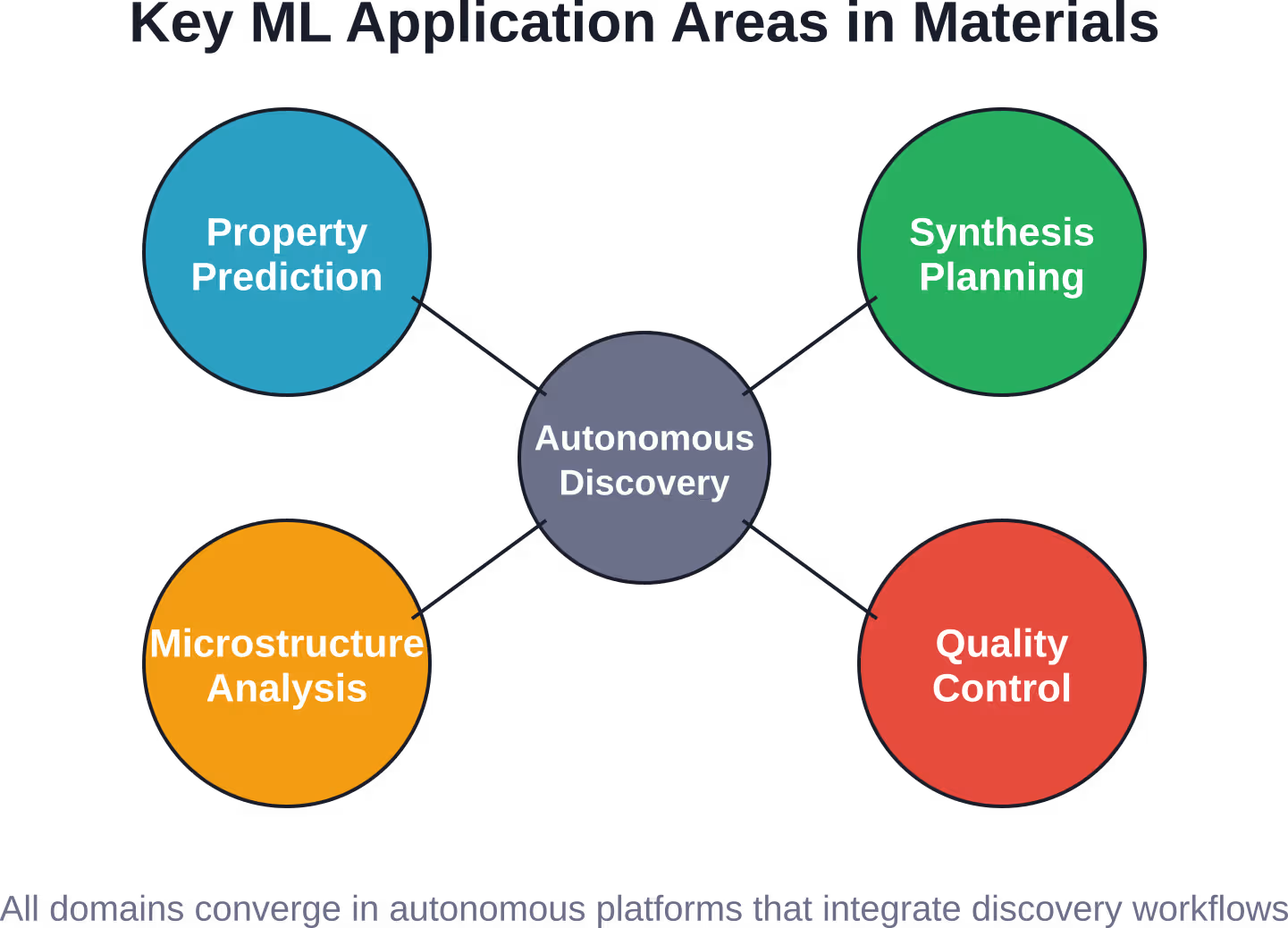
Data Challenges and Solutions
For all its promise, machine learning faces significant data challenges in materials science. Understanding these limitations is as important as understanding the capabilities.
Data Scarcity for Specific Problems
Materials databases contain hundreds of thousands of compounds, but they’re unevenly distributed. Common structural classes are well-represented; exotic compositions and structures are sparse. This creates blind spots where models perform poorly.
Active learning addresses scarcity strategically. Instead of random sampling, the algorithm identifies which experiments would be most informative—where predictions are uncertain or where new data would most improve the model. Synthesizing those materials first maximizes information gain per experiment.
Data augmentation provides another tool. Symmetry operations generate additional training examples from crystal structures. Noise injection and perturbations make models robust. These techniques artificially expand training sets, though they’re no substitute for genuine experimental diversity.
Data Quality and Standardization
Materials data comes from diverse sources using different measurement protocols, instruments, and reporting conventions. Integrating this heterogeneous data requires careful standardization and quality control.
NIST’s work setting standards for data-driven materials science addresses exactly these issues. Without agreed-upon formats, metadata standards, and quality metrics, even large datasets can be unreliable for training ML models.
Errors in training data propagate to model predictions. A mislabeled crystal structure or incorrect property measurement teaches the model the wrong relationship. Robust data curation, outlier detection, and validation against physical constraints help catch these issues before they undermine models.
The Cold Start Problem
New materials systems often lack the data needed to train accurate models. This cold start problem limits ML’s applicability for truly novel chemistries or structures.
Transfer learning from related systems provides one solution. A model trained on oxides can be fine-tuned for sulfides using limited data because fundamental chemical principles transfer. Physics-informed neural networks that encode known relationships require less data to achieve good performance because they start from realistic priors rather than blank slates.
Future Directions and Emerging Trends
The field is evolving rapidly. Several trends point toward where machine learning in materials science is headed.
Foundation Models for Materials
Large language models like GPT demonstrated that massive pre-training on diverse data creates general-purpose capabilities. Materials researchers are exploring analogous foundation models trained on comprehensive materials data—all known crystal structures, all published properties, all synthesis recipes.
These models would capture broad materials knowledge and adapt to specific tasks with minimal additional training. Early work shows promise: self-supervised pre-training improved property prediction accuracy by 6.67%, and that’s with relatively modest pre-training datasets and architectures.
The vision: a single model that handles property prediction, synthesis planning, structure determination, and literature analysis by learning unified representations of materials knowledge. This would democratize access to materials expertise.
Integration with Experimental Automation
Machine learning becomes exponentially more powerful when coupled with automated synthesis and characterization. CRESt demonstrates this potential, but current systems are limited to specific material classes and synthesis methods.
Expanding automation across diverse materials—from thin films to bulk ceramics to soft materials—will require new robotic platforms, but the payoff is enormous. Autonomous labs running 24/7 with intelligent experiment planning could compress decades of materials research into years.
The key bottleneck isn’t algorithms—it’s instrumentation. Building robotic systems that handle the full diversity of materials synthesis and characterization remains an engineering challenge.
Incorporating Uncertainty Quantification
Most ML models output point predictions: “this material has a band gap of 2.4 eV.” But for decision-making, uncertainty matters as much as the prediction. Is that 2.4 ± 0.1 eV or 2.4 ± 0.5 eV?
Bayesian approaches and ensemble methods provide uncertainty estimates, but they’re computationally expensive. Recent work on efficient uncertainty quantification—using dropout at test time, ensembling lightweight models, or learning probabilistic representations—makes uncertainty-aware predictions practical for materials applications.
Honest uncertainty estimates enable better experimental design. If a model predicts promising properties with high confidence, synthesize it. If predictions are uncertain, that material might not be worth immediate effort—or it might be interesting precisely because it explores unknown territory.
Skills and Training for Materials Scientists
Adopting machine learning requires materials scientists to acquire new skills. The good news: these skills are increasingly accessible, and institutional support is growing.
What Materials Scientists Need to Learn
Materials researchers don’t need to become ML experts, but they need enough understanding to apply tools effectively and avoid pitfalls. Essential skills include:
- Python programming and data manipulation (NumPy, Pandas)
- Basic statistics and linear algebra
- Understanding common ML algorithms (regression, decision trees, neural networks)
- Model evaluation and validation techniques
- Data visualization and interpretation
Domain knowledge remains crucial. A materials scientist with basic ML skills outperforms an ML expert with no materials knowledge because materials problems require physical intuition, data quality judgment, and interpretation of results in scientific context.
Available Training Resources
NIST’s annual Machine Learning for Materials Research Bootcamp provides intensive hands-on training covering Python basics through advanced techniques. Similar programs at universities and national labs are proliferating.
NSF’s NAIRR Classroom component expands AI education to broader communities, including materials science programs. Online courses, textbooks, and open-source software tutorials make self-directed learning increasingly feasible.
Collaboration is another route. Materials scientists partnering with computer scientists or data scientists can accomplish more than either group alone, combining domain expertise with technical ML skills.
Practical Considerations for Implementation
Organizations wanting to adopt ML in materials research face practical questions about infrastructure, workflows, and integration with existing processes.
Computing Infrastructure Requirements
Training large neural networks requires GPUs or specialized accelerators. Many universities now provide shared computing clusters with GPU nodes. Cloud providers offer on-demand access to powerful hardware without capital investment.
For many materials applications, though, modest resources suffice. Transfer learning and pre-trained models reduce computational demands. Random forests and gradient boosting machines run efficiently on standard workstations.
Storage and data management matter as much as compute. Materials datasets with diffraction patterns, micrographs, and spectroscopy results quickly reach terabytes. Organizing, versioning, and backing up this data requires thoughtful infrastructure.
Open Source Software Ecosystem
Materials scientists benefit from rich open-source ML libraries. Scikit-learn provides classical algorithms with clean APIs. PyTorch and TensorFlow enable deep learning. Materials-specific packages like Pymatgen, ASE (Atomic Simulation Environment), and MatMiner offer pre-built tools for common tasks.
This ecosystem lowers barriers to entry. Researchers can build sophisticated models using well-tested, documented libraries rather than coding algorithms from scratch.
Validation and Trust
For ML models to influence real decisions—what to synthesize, which materials to commercialize—they must be validated rigorously. Hold-out test sets, cross-validation, and comparison against experimental results establish performance baselines.
But validation extends beyond accuracy metrics. Models should be tested against physical constraints (do they violate conservation laws? predict impossible structures?), domain knowledge (do trends match chemical intuition?), and edge cases (how do they behave for extreme compositions?).
Building trust requires transparency. Document training data, model architecture, hyperparameters, and validation procedures. Provide uncertainty estimates. Make models reproducible. These practices, emphasized in NIST’s standards work, ensure ML predictions can be scrutinized and trusted.
Frequently Asked Questions
What is machine learning in materials science?
Machine learning in materials science applies algorithms that learn patterns from data to predict material properties, recommend synthesis routes, classify structures, and accelerate discovery. Instead of relying solely on experiments or simulations, researchers train models on existing materials data to make predictions about new candidates. These techniques range from simple regression to complex deep neural networks analyzing crystal structures, composition, and processing conditions.
How accurate are machine learning predictions for material properties?
Accuracy varies by property and model architecture. Formation energy predictions achieve approximately 9% mean absolute error when trained on 200,000 compounds. Microstructure classification using transfer learning reaches 98.3% accuracy. Synthesis success predictions hit 89% compared to 78% for human intuition. Virtual spectroscopy matches real measurements with 99% correlation. These numbers come from validated research, though performance depends heavily on training data quality and relevance to the prediction target.
Do materials scientists need programming skills to use machine learning?
Basic Python programming helps but isn’t always required. Many tools now offer user-friendly interfaces and pre-built workflows. That said, understanding Python, data manipulation libraries like Pandas, and ML frameworks such as scikit-learn significantly expands capabilities and control. NIST and NSF offer training programs specifically designed to teach these skills to materials researchers. Collaboration with data scientists is another effective approach when internal expertise is limited.
What types of materials problems are best suited for machine learning?
ML excels when large datasets exist, relationships are complex and non-linear, and exhaustive experimentation is impractical. Property prediction from composition or structure, image-based microstructure classification, synthesis condition optimization, and quality control in manufacturing are strong applications. Problems with very limited data, poorly understood physics, or where interpretability is absolutely critical may require more caution or hybrid approaches combining ML with traditional modeling.
How does explainable AI help materials scientists?
Explainable AI (XAI) methods reveal which features drive predictions, helping scientists understand not just what the model predicts but why. Techniques like SHAP values identify important elements or structural features. Attention mechanisms highlight relevant atoms or bonds. These insights build scientific understanding, suggest new hypotheses, and increase trust in model predictions. XAI is especially valuable when models guide expensive experiments or inform theoretical work where mechanistic understanding matters.
What data sources are available for training materials ML models?
Major databases include The Materials Project (calculated properties for over 100,000 compounds), AFLOW (crystallographic and thermodynamic data), NIST repositories (experimental measurements and standards), and published literature. Many institutions share datasets from specific studies. Data quality and standardization vary significantly across sources, so curation and validation are essential steps before training models. NIST has published guidance on data standards to address these challenges.
Can machine learning replace traditional materials experiments?
No, ML complements rather than replaces experiments. Models predict which candidates are most promising, narrowing experimental search spaces from thousands to dozens of materials. But predictions have uncertainty, and real materials exhibit complexities—defects, interfaces, processing history—that pure composition or structure don’t fully capture. The most effective approach combines ML screening with targeted validation experiments, creating an iterative cycle where predictions guide experiments and experimental results refine models.
Conclusion
Machine learning has moved from novelty to necessity in materials science. The numbers tell the story: 89% synthesis success rates beating human intuition, 98% accuracy in microstructure classification, formation energy predictions with 9% error across 20,000 test compounds, and virtual spectroscopy matching real instruments at 99% fidelity.
But the real transformation isn’t just accuracy—it’s speed and scale. Researchers now screen thousands of candidates in hours, predict properties before synthesis, and close the loop between computation and experiment through autonomous platforms. Problems that consumed decades of research compress into months or years.
Government investments totaling $100 million demonstrate recognition that materials ML is strategically important for economic competitiveness and innovation. NIST’s infrastructure and standards work ensures these techniques become reliable industrial tools, not just academic exercises.
Challenges remain. Data scarcity in new chemical spaces, the tension between accuracy and interpretability, integration with diverse experimental workflows, and training the next generation of materials scientists with hybrid expertise all require continued attention.
Yet the trajectory is clear. Machine learning is fundamentally changing how materials science operates—from reactive testing of candidates to proactive design, from experience-based intuition to data-driven predictions, from sequential workflows to closed-loop automation. The materials of 2030 will be discovered, optimized, and deployed using methods barely imaginable a decade ago.
For materials scientists, the question isn’t whether to engage with ML but how quickly and effectively to integrate it into research programs. The tools are increasingly accessible, training resources are expanding, and collaborative opportunities are growing. Organizations that build these capabilities now position themselves to lead in the next era of materials innovation.
Ready to accelerate your materials research with machine learning? Start by exploring NIST’s training programs, investigating open-source materials ML libraries, and identifying high-value prediction problems in your domain where existing data could train initial models. The infrastructure, knowledge, and community support are available—the next breakthrough material might be hiding in data you already have.