Resumen rápido: El aprendizaje automático está transformando la ciencia de los materiales al acelerar el descubrimiento, predecir propiedades y optimizar diseños que antes requerían años de desarrollo. Los investigadores ahora entrenan algoritmos en vastas bases de datos de materiales para predecir energías de formación, recomendar rutas de síntesis y clasificar microestructuras con una precisión superior al 981%. Esta revolución computacional, respaldada por iniciativas gubernamentales como una subvención de 100 millones de dólares de la NSF, en colaboración con Capital One, y la inversión de Intel en institutos de investigación de IA, permite a los científicos analizar miles de materiales candidatos en cuestión de horas, lo que cambia radicalmente la forma en que desarrollamos desde baterías hasta aleaciones estructurales.
La ciencia de los materiales siempre ha sido un proceso lento. Descubrir una nueva aleación, cerámica o polímero tradicionalmente implicaba años de experimentos de ensayo y error, costosas series de fabricación y una caracterización minuciosa. Pero ese plazo está cambiando.
Los algoritmos de aprendizaje automático ahora analizan décadas de datos de investigación en segundos, predicen las propiedades de los materiales antes de la síntesis y recomiendan candidatos prometedores entre millones de posibilidades. Según el Instituto Nacional de Estándares y Tecnología (NIST), estos flujos de trabajo impulsados por IA están demostrando un desarrollo acelerado de materiales, desde el descubrimiento hasta la comercialización e incluso la economía circular.
El cambio no es sutil. Donde la intuición humana recomendaba rutas de síntesis exitosas para materiales híbridos inorgánicos-orgánicos en el 78% de los casos, un modelo de máquina de vectores de soporte (SVM) alcanzó una precisión del 89% (89%). Ese salto de once puntos representa innumerables experimentos ahorrados y rutas más rápidas hacia el mercado.
La base: Por qué el aprendizaje automático funciona para los materiales
La ciencia de los materiales genera enormes conjuntos de datos. Cada experimento produce mediciones: patrones de difracción, resultados de espectroscopia, propiedades mecánicas, comportamiento térmico. Los investigadores han acumulado millones de estas observaciones a lo largo de décadas.
El aprendizaje automático se nutre precisamente de esta situación: grandes volúmenes de datos estructurados con relaciones complejas y no lineales. La composición atómica de un material determina su estructura cristalina, que influye en sus propiedades electrónicas, las cuales afectan su comportamiento mecánico. Estas conexiones son demasiado complejas para ecuaciones simples, pero perfectas para redes neuronales.
Sin embargo, hay algo importante: los datos de materiales son especialmente adecuados para el aprendizaje automático porque la física subyacente limita el espacio de soluciones. A diferencia de la predicción de precios de acciones o tendencias sociales, los materiales obedecen a leyes de conservación, principios termodinámicos y mecánica cuántica. Los modelos de aprendizaje automático aprenden estos patrones implícitamente a partir de los datos.
La Fundación Nacional de Ciencias (NSF) ha invertido en investigación sobre inteligencia artificial desde principios de la década de 1960, sentando las bases técnicas que impulsan las innovaciones actuales. El 29 de julio de 2025, la NSF, en colaboración con Capital One e Intel, anunció una inversión de 100 millones de dólares para apoyar a cinco Institutos Nacionales de Investigación en Inteligencia Artificial, incluido el Instituto de Materiales de IA de la NSF (NSF AI-MI), dirigido por la Universidad de Cornell.
La ventaja de los datos en la investigación de materiales
Las bases de datos de materiales han crecido exponencialmente. El Proyecto de Materiales, AFLOW y los repositorios del propio NIST contienen datos calculados y experimentales de cientos de miles de compuestos. Esta magnitud permite entrenar modelos sofisticados.
Consideremos la entalpía de formación: la energía liberada o absorbida cuando un compuesto se forma a partir de sus elementos. Una red neuronal profunda llamada ElemNet, entrenada con aproximadamente 2 × 10⁵ compuestos, logró un error absoluto medio de tan solo 0,050 ± 0,0007 eV/átomo al ser probada con aproximadamente 2 × 10⁴ compuestos diferentes. Este rendimiento permite a los investigadores seleccionar candidatos rápidamente.
La arquitectura es fundamental. ElemNet utiliza 17 capas para extraer características progresivamente abstractas de composiciones elementales básicas. Las primeras capas pueden reconocer diferencias de electronegatividad, mientras que las capas más profundas capturan tendencias de enlace complejas. Este aprendizaje jerárquico refleja la forma en que los científicos de materiales conciben las relaciones entre estructura y propiedades.
Aplicaciones clave que transforman el desarrollo de materiales
El aprendizaje automático ha pasado de ser una curiosidad académica a una herramienta de producción en múltiples ámbitos de los materiales. Sus aplicaciones se dividen en varias categorías, cada una con un impacto cuantificable.
Predicción de propiedades antes de la síntesis
La propuesta de valor más inmediata: predecir si un material tendrá las propiedades deseadas antes de invertir tiempo y dinero en su fabricación. Las redes neuronales entrenadas con cálculos de la teoría del funcional de la densidad (DFT) pueden estimar brechas de banda, módulos elásticos, conductividad térmica y docenas de otras propiedades a partir únicamente de la fórmula química.
Esta capacidad comprime el ciclo de descubrimiento. En lugar de sintetizar 100 candidatos para encontrar uno con la combinación adecuada de resistencia y conductividad, los investigadores analizan computacionalmente 10 000, sintetizan los 10 mejores y encuentran tres ganadores.
En realidad, esto no elimina la necesidad de experimentos. Las predicciones de aprendizaje automático tienen incertidumbre, y los materiales reales presentan defectos, límites de grano e historial de procesamiento que la mera composición no puede reflejar. Sin embargo, traslada el principal obstáculo experimental de la exploración general a la validación específica.
| Tipo de propiedad | Enfoque de aprendizaje automático | Precisión típica | Tamaño de los datos de entrenamiento |
|---|---|---|---|
| Energía de formación | Redes neuronales profundas | ~9% MAE | compuestos de 100.000 a 200.000 |
| Brecha de banda | Redes neuronales gráficas | MAE de 0,2–0,4 eV | 50.000–100.000 compuestos |
| Módulo de elasticidad | Bosques aleatorios | Error 10–15% | 10.000–30.000 muestras |
| Conductividad térmica | Potenciación de gradiente | Error 15–25% | Muestras de 5k a 15k |
Clasificación y análisis de la microestructura
Los científicos de materiales dedican mucho tiempo a examinar micrografías: imágenes obtenidas con microscopios ópticos, electrónicos o de fuerza atómica que revelan estructuras de grano, fases y defectos. Tradicionalmente, la clasificación de estas imágenes requería el juicio de expertos y era inherentemente subjetiva.
Las redes neuronales convolucionales (CNN) automatizan este proceso con una fidelidad extraordinaria. El aprendizaje por transferencia —que consiste en tomar una red preentrenada con millones de imágenes cotidianas y ajustarla con micrografías de materiales— logra resultados impresionantes incluso con datos de entrenamiento limitados.
El aprendizaje por transferencia con arquitecturas CNN ha logrado precisiones de clasificación de microestructuras superiores a 98% en investigaciones publicadas. No se trata de problemas de juguete; una clasificación precisa de la microestructura vincula las condiciones de procesamiento con el rendimiento, lo que permite un mejor control de calidad y una optimización del proceso.
Las implicaciones van más allá de la clasificación. Una vez que una red aprende cómo son las diferentes fases, puede cuantificar su distribución, rastrear su evolución durante el tratamiento térmico y correlacionar las características microestructurales con las propiedades mecánicas. Esto cierra el círculo entre el procesamiento, la estructura y el rendimiento.
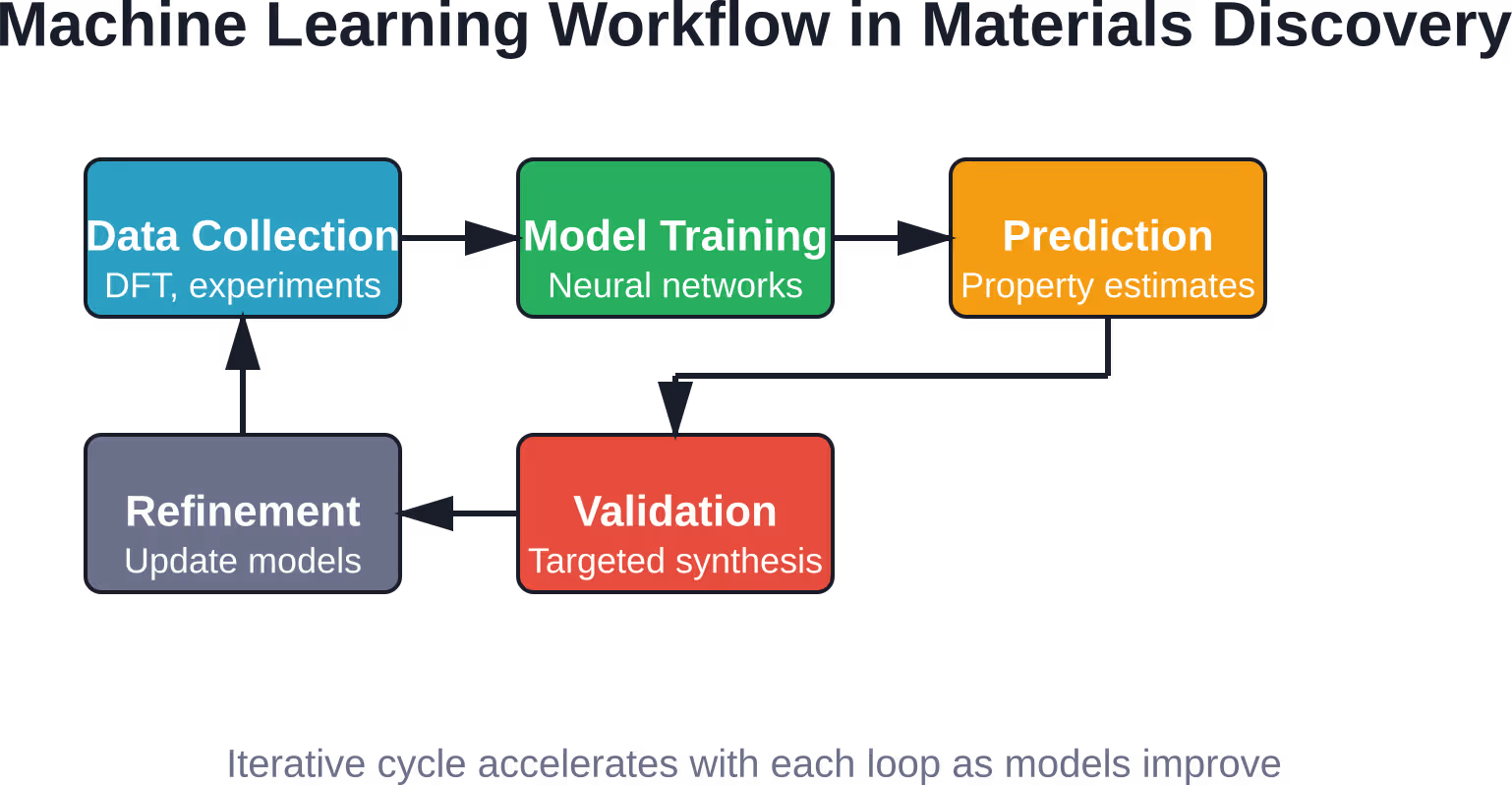
Recomendación de ruta de síntesis
Una cosa es saber qué material se desea; otra muy distinta es averiguar cómo fabricarlo. La síntesis implica elegir precursores, disolventes, temperaturas, tiempos de reacción y atmósferas. La explosión combinatoria de posibilidades es asombrosa.
Los modelos de aprendizaje automático entrenados con notas experimentales e informes de síntesis pueden recomendar rutas con alta probabilidad de éxito. Para materiales híbridos inorgánicos-orgánicos, los investigadores crearon un modelo SVM que logró una tasa de éxito en la recomendación de reacciones del 891 % en comparación con el 781 % de la intuición humana; una mejora significativa que se traduce en menos lotes fallidos y una optimización más rápida.
Estos modelos aprenden tanto de los éxitos como de los fracasos. Una reacción que no produjo la fase deseada aún proporciona información sobre qué condiciones evitar. Las técnicas de procesamiento del lenguaje natural extraen parámetros de reacción de la literatura publicada, creando automáticamente conjuntos de datos de entrenamiento.
Caracterización acelerada con espectroscopia virtual
La caracterización de materiales requiere instrumentos costosos: difractómetros de rayos X, espectrómetros infrarrojos, microscopios electrónicos. Cada modalidad proporciona información diferente, y una caracterización completa exige múltiples técnicas.
Investigadores del MIT desarrollaron SpectroGen, una herramienta de IA que funciona como un espectrómetro virtual. Al introducirle un espectro de una modalidad (por ejemplo, infrarrojo), genera el espectro que tendría ese material en otra modalidad (como rayos X). Los resultados generados por la IA coinciden con las mediciones físicas con una precisión del 991% y realizan predicciones completas en menos de un minuto.
Esta capacidad reduce drásticamente los costos y el tiempo de caracterización. Un fabricante puede realizar una medición rápida y usar SpectroGen para generar espectros predictivos en múltiples modalidades, detectando problemas de calidad o fases inesperadas sin necesidad de acceder a todos los instrumentos. Para las industrias que producen materiales a gran escala, esto representa enormes mejoras en la eficiencia.

Utilice el aprendizaje automático en la ciencia de los materiales con IA superior
La investigación en ciencia de los materiales suele generar grandes conjuntos de datos procedentes de simulaciones, entornos de prueba y experimentos de laboratorio. IA superior Pueden ayudar a los equipos a estructurar proyectos de aprendizaje automático para el análisis de materiales, el modelado predictivo y la automatización de la investigación. Su trabajo abarca consultoría en IA, ciencia de datos, ingeniería de aprendizaje automático, desarrollo de pruebas de concepto y soporte de software de IA.
AI Superior puede ayudar a los proyectos de ciencia de materiales a través de:
- Preparación y evaluación de conjuntos de datos de materiales
- Desarrollo de modelos de aprendizaje automático para el análisis de materiales.
- Creación de flujos de trabajo predictivos para entornos de investigación
- Detección de irregularidades y patrones de comportamiento del material
- Validación y prueba de modelos analíticos
- Integración de la planificación en los sistemas de investigación internos
En el ámbito de la ciencia de los materiales, esto puede incluir el análisis de defectos, la predicción de propiedades de los materiales, el apoyo a la simulación y el procesamiento de datos experimentales.
👉Contacta con IA Superior para explorar la configuración del proyecto y los próximos pasos.
Arquitecturas de aprendizaje profundo para problemas de materiales
No todos los modelos de aprendizaje automático son iguales. Las aplicaciones de materiales se han inclinado hacia arquitecturas específicas que manejan las características únicas de los datos de materiales.
Redes neuronales gráficas para estructuras cristalinas
Los cristales tienen una estructura intrínsecamente gráfica: los átomos son nodos, los enlaces son aristas y la topología de la red codifica la estructura. Las redes neuronales gráficas (GNN) operan directamente sobre esta representación, lo que las hace idóneas para los materiales cristalinos.
Una red neuronal gráfica (GNN) procesa un cristal actualizando iterativamente la representación de cada átomo en función de sus vecinos. Tras varias rondas de intercambio de mensajes, la red construye una representación que captura los entornos de enlace locales, los motivos estructurales de alcance medio y la simetría global, todos ellos relevantes para las propiedades del cristal.
Las redes neuronales gráficas (GNN) han demostrado una gran capacidad para predecir propiedades relacionadas con la estructura electrónica y los enlaces químicos. Superan a los modelos tradicionales basados en descriptores para brechas de banda, energías de formación y propiedades magnéticas, ya que codifican directamente las relaciones geométricas y químicas en lugar de depender de características diseñadas manualmente.
Estrategias de aprendizaje por transferencia y preformación
Si bien los conjuntos de datos de materiales son extensos para los estándares científicos, resultan pequeños en comparación con los millones de imágenes o los miles de millones de tokens de texto que se utilizan para entrenar la IA de propósito general. El aprendizaje por transferencia aborda esta brecha.
Una red neuronal preentrenada con un conjunto de datos amplio y general aprende características de gran utilidad, como la detección de bordes en imágenes o las relaciones químicas generales en moléculas. El ajuste fino de esta red con un conjunto de datos de materiales más pequeño y especializado adapta dichas características a la tarea específica.
El preentrenamiento auto-supervisado ofrece otra alternativa. Un modelo aprende prediciendo propiedades ocultas o reconstruyendo estructuras dañadas. Un estudio publicado en Nature demostró que el preentrenamiento auto-supervisado mejoró el rendimiento en la predicción de propiedades de materiales en 6,67% en términos de error absoluto medio (MAE).
Aprendizaje multitarea y multifidelidad
Las propiedades de los materiales están correlacionadas. Un material con un punto de fusión alto suele tener una dureza elevada. La conductividad térmica y eléctrica suelen ir de la mano en los metales. El aprendizaje multitarea aprovecha estas correlaciones entrenando un único modelo para predecir múltiples propiedades simultáneamente.
La representación compartida, aprendida a través de diversas tareas, captura los factores químicos y estructurales subyacentes que influyen en todas las propiedades. Este enfoque suele superar a los modelos independientes para cada tarea, especialmente cuando los datos de entrenamiento para algunas propiedades son escasos, ya que el modelo aprovecha la información estadística de tareas relacionadas.
El aprendizaje multifidelidad aborda otro desafío importante: la combinación de datos de diferentes fuentes. Los cálculos DFT de alta fidelidad son precisos pero costosos; los modelos empíricos son rápidos pero aproximados. Un modelo multifidelidad aprende a utilizar datos económicos de baja fidelidad para corregir sesgos y complementar los costosos datos de alta fidelidad, maximizando así la extracción de información de los recursos disponibles.
El desafío de la explicabilidad y las soluciones de la IA explicable
El problema radica en que los modelos de aprendizaje automático más precisos suelen ser cajas negras. Una red neuronal profunda con millones de parámetros puede predecir las propiedades de los materiales con gran exactitud, pero no explica el porqué. Para los investigadores que intentan comprender los principios físicos, esto resulta frustrante.
La inteligencia artificial explicable (XAI) aborda este problema. El objetivo no son solo predicciones precisas, sino predicciones interpretables que revelen información química y contribuyan al conocimiento científico.
Qué significa “explicar” en el contexto de los materiales.
La explicación adopta diferentes formas según el público y la aplicación. Para un investigador de síntesis, la explicación podría significar identificar qué parámetros de reacción influyen más en el rendimiento. Para un teórico, podría significar revelar qué características de la estructura electrónica determinan la estabilidad.
Los métodos de importancia de características clasifican las variables de entrada según su contribución a las predicciones. Los valores SHAP (SHapley Additive exPlanations), derivados de la teoría de juegos, proporcionan una forma sistemática de asignar el mérito a cada característica de entrada para cada predicción. Si un modelo predice una alta conductividad, SHAP revela qué elementos y características estructurales impulsaron dicha predicción.
Los mecanismos de atención en las redes neuronales ofrecen otra vía para la interpretabilidad. El modelo aprende explícitamente qué partes de la entrada (qué átomos, qué enlaces) son relevantes para cada propiedad. La visualización de estos pesos de atención resalta los motivos estructurales que controlan el comportamiento.
Equilibrar la precisión y la interpretabilidad
Existe una tensión entre precisión e interpretabilidad. Los modelos lineales son transparentes, pero a menudo imprecisos. Las redes neuronales profundas son precisas, pero opacas. La solución práctica suele implicar un compromiso.
Una estrategia consiste en utilizar un modelo complejo para las predicciones, pero ajustar un modelo más simple e interpretable para aproximar su comportamiento localmente. LIME (Local Interpretable Model-agnostic Explanations) implementa esta idea, construyendo aproximaciones lineales locales en torno a predicciones individuales. La aproximación explica esa predicción específica, incluso si el modelo subyacente es complejo.
Otro enfoque incorpora la interpretabilidad a la arquitectura. Las redes neuronales con capas especializadas que codifican principios físicos conocidos (leyes de conservación, restricciones de simetría, descriptores específicos del dominio) son más precisas y más interpretables que las arquitecturas genéricas, ya que su estructura refleja conceptos reales de la ciencia de los materiales.
| Método XAI | Tipo de interpretación | Mejor para | Limitación |
|---|---|---|---|
| Valores SHAP | Importancia de la característica | Comprender a los conductores | Computacionalmente costoso |
| Visualización de la atención | Motivos estructurales | Identificación de características clave | Específico de la arquitectura |
| CAL | aproximación lineal local | predicciones individuales | Solo validez local |
| Mapas de prominencia | Sensibilidad de entrada | datos de imagen/estructura | Puede ser ruidoso |
Iniciativas gubernamentales e institucionales que impulsan el progreso
El aprendizaje automático en la ciencia de los materiales no es solo investigación académica, sino una prioridad estratégica para los gobiernos y las principales instituciones, que reconocen sus implicaciones económicas y de seguridad.
Esfuerzos del NIST en ciencia de materiales basados en datos e inteligencia artificial
El Instituto Nacional de Estándares y Tecnología ha creado un Grupo de Ciencia de Materiales impulsado por Datos e Inteligencia Artificial dedicado al desarrollo de métodos, algoritmos, datos y herramientas para acelerar el descubrimiento, el desarrollo, la comercialización y la circularidad de materiales de relevancia industrial.
El NIST organiza anualmente un curso intensivo de aprendizaje automático para la investigación de materiales, que consta de cuatro días de conferencias y ejercicios prácticos. Los temas abarcan desde los fundamentos de Python y el preprocesamiento de datos hasta técnicas avanzadas de aprendizaje automático, proporcionando una formación práctica que capacita a los investigadores para aplicar estos métodos.
Establecer estándares es fundamental. Investigadores del NIST publicaron recientemente una guía sobre estándares para la ciencia de materiales basada en datos, abordando la calidad de los datos, la validación de modelos y la reproducibilidad: cuestiones fundamentales que deben resolverse para que el aprendizaje automático ofrezca aplicaciones industriales fiables.
Infraestructura nacional de investigación en IA de la NSF
La Fundación Nacional de Ciencias (NSF) lidera el Recurso Nacional de Investigación en Inteligencia Artificial (NAIRR, por sus siglas en inglés), una infraestructura nacional que proporciona a las comunidades de investigación y educación acceso a la computación, el software, los datos, los modelos y la experiencia necesarios para la innovación en IA.
NAIRR se centra en ampliar el acceso a los recursos de IA en toda la comunidad investigadora, incluidos los científicos de materiales. El componente NAIRR Classroom de la NSF desarrolla capacidades de personal preparadas para la IA mediante educación, capacitación y divulgación a comunidades nuevas y no tradicionales.
Este enfoque de infraestructura reconoce que la investigación de vanguardia en aprendizaje automático requiere recursos computacionales que van más allá de los que suelen proporcionar las universidades individuales. Democratizar el acceso garantiza que la innovación en materiales no se limite a un puñado de instituciones con gran financiación.
Colaboración y competencia internacionales
La ciencia de los materiales es, por naturaleza, internacional. La Iniciativa del Genoma de los Materiales en Estados Unidos es paralela a iniciativas similares en Europa, Japón y China. El aprendizaje automático se ha convertido en una ventaja competitiva en este ámbito.
Los países que desarrollan bases de datos de materiales superiores, forman a más científicos de materiales con capacidad para la IA y mejoran su infraestructura computacional obtienen ventajas en industrias que van desde la aeroespacial hasta la electrónica y la energética. La inversión de 100 millones de dólares del programa $, anunciada el 29 de julio de 2025, tiene como objetivo explícito fortalecer la competitividad global de Estados Unidos y acelerar la innovación.
Aplicaciones prácticas y estudios de caso
Basta de teoría. ¿Qué están haciendo realmente las organizaciones con estas técnicas?
Optimización de materiales para baterías
Las empresas de baterías necesitan comprender cómo los parámetros de fabricación afectan el rendimiento de las celdas. Los modelos de aprendizaje automático representan estas relaciones, lo que permite la optimización para aplicaciones específicas, la reducción de costos y la mejora del rendimiento.
Investigadores de Stanford y otras instituciones aplican el aprendizaje automático para caracterizar y diseñar electrodos de baterías, analizando cómo la composición, la distribución del tamaño de partícula, la porosidad y el contenido de aglutinante influyen en la capacidad, la velocidad de carga/descarga y la vida útil. Estos modelos aceleran el proceso de diseño iterativo que tradicionalmente requería cientos de lotes experimentales.
Plataformas autónomas para el descubrimiento de materiales
La plataforma CRESt (Tecnología de Búsqueda Experimental Robótica de Bucle Cerrado) del MIT representa la siguiente evolución: el descubrimiento totalmente autónomo. El sistema combina el aprendizaje automático con la síntesis y caracterización robótica para ejecutar experimentos, analizar resultados, actualizar modelos y diseñar los siguientes experimentos, todo ello sin intervención humana.
CRESt aprende de diversas fuentes científicas —literatura, bases de datos, resultados experimentales— y genera soluciones a problemas energéticos que han afectado a la ciencia de los materiales durante décadas. Este enfoque de ciclo cerrado descubre materiales a una velocidad mucho mayor que la investigación guiada por humanos.
El sistema no solo predice, sino que explora activamente, equilibrando la explotación (sintetizando materiales que predice que funcionarán) con la exploración (probando candidatos inciertos que podrían revelar nuevos conocimientos). Esta estrategia, tomada del aprendizaje por refuerzo, navega eficientemente por vastos espacios de búsqueda.
Control de calidad en la fabricación
La producción de materiales industriales requiere una calidad constante. Los modelos de aprendizaje automático supervisan los datos de los sensores en tiempo real durante la fabricación, prediciendo las propiedades y detectando las desviaciones antes de que se conviertan en fallos costosos.
Una aplicación predice la dureza de metales de baja aleación a partir de su composición y parámetros de procesamiento. En lugar de esperar a las pruebas posteriores a la producción, el modelo proporciona información instantánea, lo que permite realizar ajustes en el proceso para mantener la producción dentro de las especificaciones.
La espectroscopia virtual de SpectroGen encuentra aquí una aplicación natural. Una única medición rápida, seguida de espectros multimodales generados por IA, proporciona una evaluación de calidad integral en menos de un minuto, lo suficientemente rápida para su integración en la línea de producción.
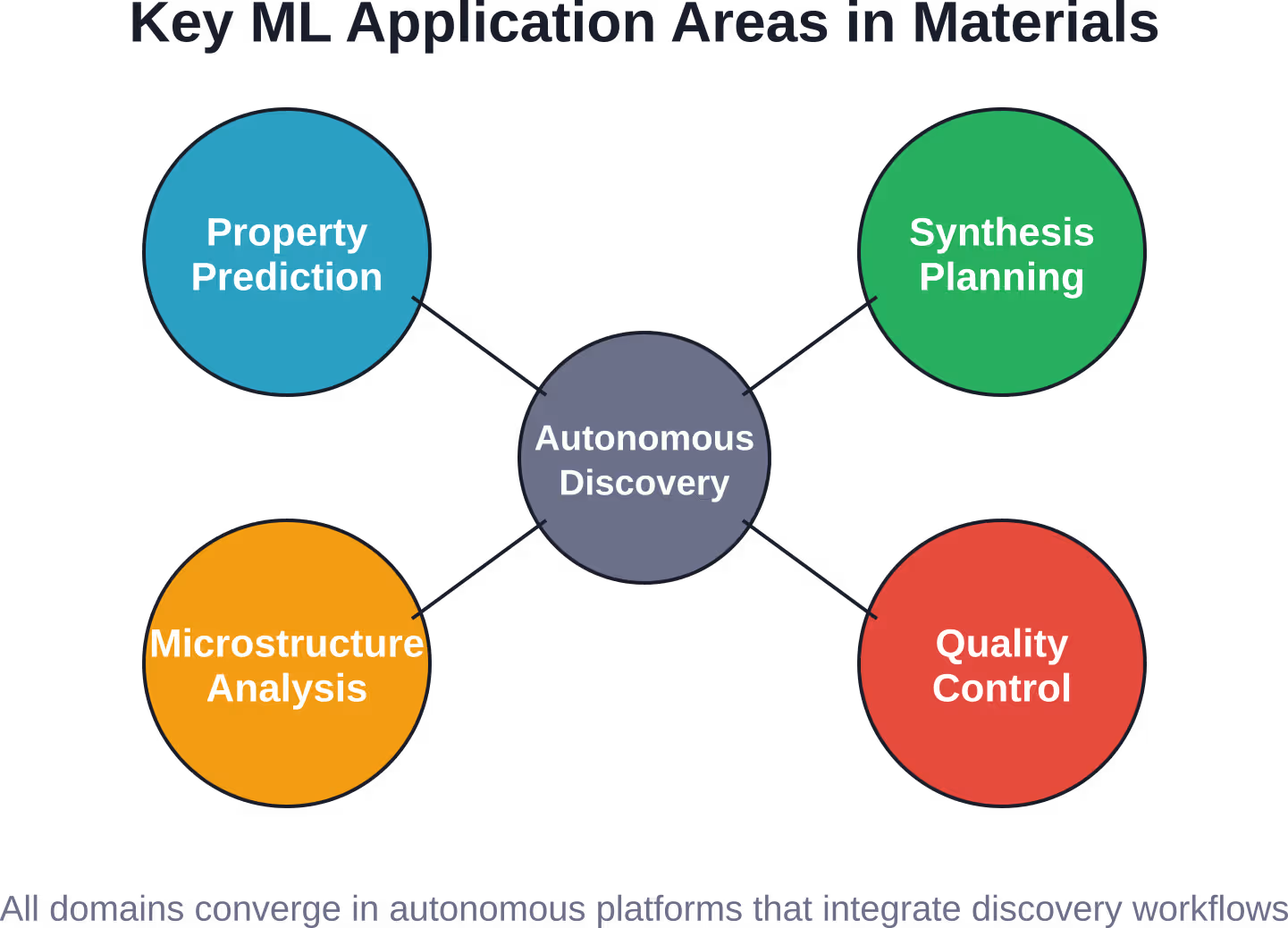
Desafíos y soluciones relacionados con los datos
A pesar de su gran potencial, el aprendizaje automático se enfrenta a importantes desafíos en el manejo de datos en la ciencia de los materiales. Comprender estas limitaciones es tan importante como comprender sus capacidades.
Escasez de datos para problemas específicos
Las bases de datos de materiales contienen cientos de miles de compuestos, pero su distribución es desigual. Las clases estructurales comunes están bien representadas; las composiciones y estructuras exóticas son escasas. Esto crea puntos ciegos donde los modelos tienen un rendimiento deficiente.
El aprendizaje activo aborda la escasez de forma estratégica. En lugar de un muestreo aleatorio, el algoritmo identifica qué experimentos serían los más informativos: dónde las predicciones son inciertas o dónde los nuevos datos mejorarían más el modelo. Sintetizar primero esos materiales maximiza la ganancia de información por experimento.
El aumento de datos proporciona otra herramienta. Las operaciones de simetría generan ejemplos de entrenamiento adicionales a partir de estructuras cristalinas. La inyección de ruido y las perturbaciones hacen que los modelos sean robustos. Estas técnicas expanden artificialmente los conjuntos de entrenamiento, aunque no sustituyen la diversidad experimental genuina.
Calidad y estandarización de los datos
Los datos sobre materiales provienen de diversas fuentes que utilizan diferentes protocolos de medición, instrumentos y convenciones de presentación de informes. La integración de estos datos heterogéneos requiere una estandarización y un control de calidad rigurosos.
El trabajo del NIST para establecer estándares en la ciencia de los materiales basada en datos aborda precisamente estos problemas. Sin formatos, estándares de metadatos y métricas de calidad consensuados, incluso los conjuntos de datos grandes pueden resultar poco fiables para el entrenamiento de modelos de aprendizaje automático.
Los errores en los datos de entrenamiento se propagan a las predicciones del modelo. Una estructura cristalina mal etiquetada o una medición de propiedad incorrecta enseñan al modelo una relación errónea. Una gestión rigurosa de los datos, la detección de valores atípicos y la validación con respecto a restricciones físicas ayudan a detectar estos problemas antes de que afecten negativamente a los modelos.
El problema del arranque en frío
Los sistemas de materiales nuevos a menudo carecen de los datos necesarios para entrenar modelos precisos. Este problema inicial limita la aplicabilidad del aprendizaje automático a químicas o estructuras verdaderamente novedosas.
El aprendizaje por transferencia a partir de sistemas relacionados ofrece una solución. Un modelo entrenado con óxidos puede ajustarse para sulfuros con datos limitados, ya que los principios químicos fundamentales son transferibles. Las redes neuronales basadas en la física que codifican relaciones conocidas requieren menos datos para lograr un buen rendimiento, puesto que parten de información previa realista en lugar de partir de cero.
Direcciones futuras y tendencias emergentes
Este campo está evolucionando rápidamente. Varias tendencias apuntan hacia dónde se dirige el aprendizaje automático en la ciencia de los materiales.
Modelos fundamentales para materiales
Los modelos de lenguaje a gran escala, como GPT, demostraron que el preentrenamiento masivo con datos diversos genera capacidades de propósito general. Los investigadores de materiales están explorando modelos base análogos, entrenados con datos exhaustivos de materiales: todas las estructuras cristalinas conocidas, todas las propiedades publicadas y todas las recetas de síntesis.
Estos modelos capturarían un amplio conocimiento de los materiales y se adaptarían a tareas específicas con un mínimo de entrenamiento adicional. Los primeros trabajos son prometedores: el preentrenamiento auto-supervisado mejoró la precisión de la predicción de propiedades en 6,67%, y eso con conjuntos de datos y arquitecturas de preentrenamiento relativamente modestos.
La visión: un modelo único que gestione la predicción de propiedades, la planificación de síntesis, la determinación de estructuras y el análisis bibliográfico mediante el aprendizaje de representaciones unificadas del conocimiento de los materiales. Esto democratizaría el acceso a la experiencia en materiales.
Integración con la automatización experimental
El aprendizaje automático se vuelve exponencialmente más potente cuando se combina con la síntesis y caracterización automatizadas. CRESt demuestra este potencial, pero los sistemas actuales se limitan a clases de materiales y métodos de síntesis específicos.
Ampliar la automatización a diversos materiales —desde películas delgadas hasta cerámicas a granel y materiales blandos— requerirá nuevas plataformas robóticas, pero la recompensa es enorme. Los laboratorios autónomos que funcionen las 24 horas del día, los 7 días de la semana, con planificación inteligente de experimentos, podrían condensar décadas de investigación de materiales en solo unos años.
El principal obstáculo no son los algoritmos, sino la instrumentación. Construir sistemas robóticos capaces de gestionar toda la diversidad de síntesis y caracterización de materiales sigue siendo un reto de ingeniería.
Incorporación de la cuantificación de la incertidumbre
La mayoría de los modelos de aprendizaje automático generan predicciones puntuales: “este material tiene una banda prohibida de 2,4 eV”. Pero para la toma de decisiones, la incertidumbre es tan importante como la predicción. ¿Se trata de 2,4 ± 0,1 eV o de 2,4 ± 0,5 eV?
Los enfoques bayesianos y los métodos de conjunto proporcionan estimaciones de incertidumbre, pero son computacionalmente costosos. Trabajos recientes sobre la cuantificación eficiente de la incertidumbre —mediante el uso de abandono en el momento de la prueba, la combinación de modelos ligeros o el aprendizaje de representaciones probabilísticas— hacen que las predicciones que tienen en cuenta la incertidumbre sean prácticas para aplicaciones de materiales.
Las estimaciones honestas de la incertidumbre permiten un mejor diseño experimental. Si un modelo predice propiedades prometedoras con alta confianza, conviene sintetizarlo. Si las predicciones son inciertas, ese material podría no merecer un esfuerzo inmediato, o podría ser interesante precisamente porque explora un territorio desconocido.
Habilidades y formación para científicos de materiales
La adopción del aprendizaje automático exige que los científicos de materiales adquieran nuevas habilidades. La buena noticia es que estas habilidades son cada vez más accesibles y el apoyo institucional está creciendo.
Lo que los científicos de materiales necesitan aprender
Los investigadores de materiales no necesitan convertirse en expertos en aprendizaje automático, pero sí necesitan comprender lo suficiente para aplicar las herramientas de manera efectiva y evitar errores. Las habilidades esenciales incluyen:
- Programación en Python y manipulación de datos (NumPy, Pandas)
- Estadística básica y álgebra lineal
- Comprensión de los algoritmos comunes de aprendizaje automático (regresión, árboles de decisión, redes neuronales)
- Técnicas de evaluación y validación de modelos
- Visualización e interpretación de datos
El conocimiento del dominio sigue siendo crucial. Un científico de materiales con habilidades básicas de aprendizaje automático supera a un experto en aprendizaje automático sin conocimientos de materiales, porque los problemas de materiales requieren intuición física, criterio para evaluar la calidad de los datos e interpretación de los resultados en un contexto científico.
Recursos de capacitación disponibles
El programa anual de capacitación intensiva en aprendizaje automático para la investigación de materiales del NIST ofrece formación práctica que abarca desde los fundamentos de Python hasta técnicas avanzadas. Programas similares en universidades y laboratorios nacionales están proliferando.
El componente NAIRR Classroom de la NSF amplía la educación en IA a comunidades más amplias, incluidos los programas de ciencia de los materiales. Los cursos en línea, los libros de texto y los tutoriales de software de código abierto hacen que el aprendizaje autodirigido sea cada vez más factible.
La colaboración es otra vía. Los científicos de materiales que se asocian con informáticos o científicos de datos pueden lograr más que cualquiera de los grupos por separado, combinando la experiencia en el campo con habilidades técnicas de aprendizaje automático.
Consideraciones prácticas para la implementación
Las organizaciones que desean adoptar el aprendizaje automático en la investigación de materiales se enfrentan a cuestiones prácticas sobre infraestructura, flujos de trabajo e integración con los procesos existentes.
Requisitos de infraestructura informática
El entrenamiento de grandes redes neuronales requiere GPU o aceleradores especializados. Muchas universidades ofrecen ahora clústeres de computación compartida con nodos GPU. Los proveedores de servicios en la nube ofrecen acceso bajo demanda a hardware potente sin necesidad de inversión inicial.
Sin embargo, para muchas aplicaciones de materiales, bastan recursos modestos. El aprendizaje por transferencia y los modelos preentrenados reducen las exigencias computacionales. Los bosques aleatorios y las máquinas de potenciación de gradiente funcionan de manera eficiente en estaciones de trabajo estándar.
El almacenamiento y la gestión de datos son tan importantes como la capacidad de procesamiento. Los conjuntos de datos de materiales, que incluyen patrones de difracción, micrografías y resultados de espectroscopia, alcanzan rápidamente los terabytes. Organizar, versionar y realizar copias de seguridad de estos datos requiere una infraestructura bien planificada.
Ecosistema de software de código abierto
Los científicos de materiales se benefician de una amplia gama de bibliotecas de aprendizaje automático de código abierto. Scikit-learn proporciona algoritmos clásicos con API intuitivas. PyTorch y TensorFlow permiten el aprendizaje profundo. Paquetes específicos para materiales como Pymatgen, ASE (Atomic Simulation Environment) y MatMiner ofrecen herramientas preconfiguradas para tareas comunes.
Este ecosistema reduce las barreras de entrada. Los investigadores pueden crear modelos sofisticados utilizando bibliotecas bien probadas y documentadas, en lugar de programar algoritmos desde cero.
Validación y confianza
Para que los modelos de aprendizaje automático influyan en decisiones reales —qué sintetizar, qué materiales comercializar— deben validarse rigurosamente. Los conjuntos de prueba independientes, la validación cruzada y la comparación con resultados experimentales establecen parámetros de referencia de rendimiento.
Pero la validación va más allá de las métricas de precisión. Los modelos deben someterse a pruebas con respecto a restricciones físicas (¿violan las leyes de conservación? ¿predicen estructuras imposibles?), conocimiento del dominio (¿coinciden las tendencias con la intuición química?) y casos extremos (¿cómo se comportan para composiciones extremas?).
Generar confianza requiere transparencia. Documente los datos de entrenamiento, la arquitectura del modelo, los hiperparámetros y los procedimientos de validación. Proporcione estimaciones de incertidumbre. Asegúrese de que los modelos sean reproducibles. Estas prácticas, destacadas en los estándares del NIST, garantizan que las predicciones de aprendizaje automático puedan ser examinadas y consideradas fiables.
Preguntas frecuentes
¿Qué es el aprendizaje automático en la ciencia de los materiales?
El aprendizaje automático en la ciencia de los materiales aplica algoritmos que aprenden patrones a partir de datos para predecir propiedades de los materiales, recomendar rutas de síntesis, clasificar estructuras y acelerar el descubrimiento. En lugar de depender únicamente de experimentos o simulaciones, los investigadores entrenan modelos con datos de materiales existentes para realizar predicciones sobre nuevos candidatos. Estas técnicas abarcan desde la regresión simple hasta complejas redes neuronales profundas que analizan estructuras cristalinas, composición y condiciones de procesamiento.
¿Qué tan precisas son las predicciones de aprendizaje automático sobre las propiedades de los materiales?
La precisión varía según la propiedad y la arquitectura del modelo. Las predicciones de energía de formación alcanzan un error absoluto medio de aproximadamente 91 TP3T al entrenarse con 200 000 compuestos. La clasificación de microestructura mediante aprendizaje por transferencia alcanza una precisión de 98,31 TP3T. Las predicciones de éxito de síntesis alcanzan 891 TP3T en comparación con 781 TP3T para la intuición humana. La espectroscopia virtual coincide con las mediciones reales con una correlación de 991 TP3T. Estas cifras provienen de investigaciones validadas, aunque el rendimiento depende en gran medida de la calidad de los datos de entrenamiento y su relevancia para el objetivo de predicción.
¿Necesitan los científicos de materiales conocimientos de programación para utilizar el aprendizaje automático?
Tener conocimientos básicos de programación en Python es útil, pero no siempre imprescindible. Muchas herramientas ofrecen interfaces intuitivas y flujos de trabajo predefinidos. Sin embargo, comprender Python, bibliotecas de manipulación de datos como Pandas y marcos de aprendizaje automático como scikit-learn amplía significativamente las capacidades y el control. El NIST y la NSF ofrecen programas de formación diseñados específicamente para enseñar estas habilidades a los investigadores de materiales. La colaboración con científicos de datos es otra estrategia eficaz cuando la experiencia interna es limitada.
¿Qué tipos de problemas relacionados con los materiales son los más adecuados para el aprendizaje automático?
El aprendizaje automático destaca cuando se dispone de grandes conjuntos de datos, las relaciones son complejas y no lineales, y la experimentación exhaustiva resulta impracticable. La predicción de propiedades a partir de la composición o la estructura, la clasificación de microestructuras basada en imágenes, la optimización de las condiciones de síntesis y el control de calidad en la fabricación son aplicaciones importantes. Los problemas con datos muy limitados, una física poco comprendida o donde la interpretabilidad es fundamental pueden requerir mayor precaución o enfoques híbridos que combinen el aprendizaje automático con el modelado tradicional.
¿Cómo ayuda la IA explicable a los científicos de materiales?
Los métodos de IA explicable (XAI) revelan qué características impulsan las predicciones, lo que ayuda a los científicos a comprender no solo qué predice el modelo, sino también por qué. Técnicas como los valores SHAP identifican elementos o características estructurales importantes. Los mecanismos de atención resaltan átomos o enlaces relevantes. Estos conocimientos contribuyen a la comprensión científica, sugieren nuevas hipótesis y aumentan la confianza en las predicciones del modelo. La XAI es especialmente valiosa cuando los modelos guían experimentos costosos o aportan información a trabajos teóricos donde la comprensión de los mecanismos es fundamental.
¿Qué fuentes de datos están disponibles para los materiales de entrenamiento de los modelos de aprendizaje automático?
Las principales bases de datos incluyen The Materials Project (propiedades calculadas para más de 100 000 compuestos), AFLOW (datos cristalográficos y termodinámicos), los repositorios del NIST (mediciones y estándares experimentales) y la literatura publicada. Muchas instituciones comparten conjuntos de datos de estudios específicos. La calidad y la estandarización de los datos varían significativamente entre las distintas fuentes, por lo que la curación y la validación son pasos esenciales antes de entrenar los modelos. El NIST ha publicado directrices sobre estándares de datos para abordar estos desafíos.
¿Puede el aprendizaje automático sustituir los experimentos tradicionales con materiales?
No, el aprendizaje automático complementa los experimentos, no los reemplaza. Los modelos predicen qué candidatos son los más prometedores, reduciendo el espacio de búsqueda experimental de miles a decenas de materiales. Sin embargo, las predicciones tienen incertidumbre, y los materiales reales presentan complejidades —defectos, interfaces, historial de procesamiento— que la composición o estructura por sí solas no capturan por completo. El enfoque más eficaz combina la selección mediante aprendizaje automático con experimentos de validación específicos, creando un ciclo iterativo donde las predicciones guían los experimentos y los resultados experimentales refinan los modelos.
Conclusión
El aprendizaje automático ha pasado de ser una novedad a una necesidad en la ciencia de los materiales. Las cifras lo demuestran: tasas de éxito de síntesis del 891% que superan la intuición humana, precisión del 98% en la clasificación de microestructuras, predicciones de energía de formación con un margen de error del 91% en 20 000 compuestos de prueba, y espectroscopia virtual que iguala la fidelidad de los instrumentos reales al 991%.
Pero la verdadera transformación no reside solo en la precisión, sino también en la velocidad y la escala. Ahora, los investigadores analizan miles de candidatos en cuestión de horas, predicen propiedades antes de la síntesis y cierran el ciclo entre la computación y la experimentación mediante plataformas autónomas. Problemas que antes consumían décadas de investigación ahora se resuelven en meses o años.
Las inversiones gubernamentales, que ascienden a 14.000 millones de dólares, demuestran el reconocimiento de que el aprendizaje automático de materiales es estratégicamente importante para la competitividad económica y la innovación. La infraestructura y el trabajo de estandarización del NIST garantizan que estas técnicas se conviertan en herramientas industriales fiables, y no solo en ejercicios académicos.
Persisten los desafíos. La escasez de datos en nuevos ámbitos químicos, la tensión entre precisión e interpretabilidad, la integración con diversos flujos de trabajo experimentales y la formación de la próxima generación de científicos de materiales con conocimientos híbridos requieren una atención continua.
Sin embargo, la trayectoria es clara. El aprendizaje automático está transformando radicalmente el funcionamiento de la ciencia de los materiales: desde las pruebas reactivas de candidatos hasta el diseño proactivo, desde la intuición basada en la experiencia hasta las predicciones basadas en datos, desde los flujos de trabajo secuenciales hasta la automatización de ciclo cerrado. Los materiales de 2030 se descubrirán, optimizarán y utilizarán mediante métodos prácticamente inimaginables hace una década.
Para los científicos de materiales, la cuestión no es si utilizar el aprendizaje automático, sino con qué rapidez y eficacia integrarlo en los programas de investigación. Las herramientas son cada vez más accesibles, los recursos de formación se amplían y las oportunidades de colaboración crecen. Las organizaciones que desarrollan estas capacidades se posicionan ahora para liderar la próxima era de la innovación en materiales.
¿Listo para acelerar tu investigación de materiales con aprendizaje automático? Empieza explorando los programas de capacitación del NIST, investigando las bibliotecas de aprendizaje automático de materiales de código abierto e identificando problemas de predicción de alto valor en tu campo donde los datos existentes puedan entrenar los modelos iniciales. La infraestructura, el conocimiento y el apoyo de la comunidad están a tu disposición: el próximo material revolucionario podría estar oculto en los datos que ya tienes.