Resumen rápido: El aprendizaje automático está revolucionando la química al acelerar el descubrimiento de fármacos, predecir propiedades moleculares y diseñar nuevos materiales. Con algoritmos prometedores en la predicción de interacciones proteicas y la previsión de síntesis de materiales, el aprendizaje automático está transformando la investigación química tradicional, pasando del método de ensayo y error a la precisión basada en datos, lo que reduce drásticamente el tiempo y los costes de desarrollo.
La industria farmacéutica se enfrenta a una cruda realidad: las tasas de éxito en el desarrollo de fármacos rondan entre el 9,6 % y el 121 % desde los ensayos de fase I hasta la aprobación final. Los métodos tradicionales consumen años y miles de millones de dólares, pero fracasan con más frecuencia de la que tienen éxito.
El aprendizaje automático está cambiando esa situación. Al procesar enormes conjuntos de datos químicos e identificar patrones invisibles para los investigadores humanos, estos algoritmos están acelerando los plazos de descubrimiento y mejorando la precisión en múltiples ámbitos.
El descubrimiento de fármacos se renueva gracias a los datos.
Sin embargo, hay algo importante: el aprendizaje automático sobresale precisamente donde la química tradicional tiene más dificultades. El reconocimiento de patrones en vastas bibliotecas moleculares, la predicción de propiedades sin síntesis física y la identificación de objetivos se benefician de la precisión algorítmica.
Los modelos de aprendizaje profundo predicen ahora las interacciones proteína-proteína con una precisión notable. Sin embargo, el desarrollo de fármacos sigue siendo un reto. La tasa de éxito general desde los ensayos clínicos de fase I hasta la aprobación de un fármaco es de aproximadamente 9,6 a 121 TP3T, aunque varía significativamente según el área terapéutica (por ejemplo, ~31 TP3T en oncología). La brecha entre las promesas de la simulación y la realidad clínica sigue siendo considerable.
Generación molecular y predicción de propiedades
Los modelos generativos crean estructuras moleculares completamente nuevas con las propiedades deseadas. Diversos enfoques generativos muestran diferentes tasas de validez para la generación molecular. Estos no son logros triviales: generar estructuras químicamente plausibles requiere comprender las reglas de enlace, las restricciones de estabilidad y la accesibilidad sintética.
Los modelos de aprendizaje automático que utilizan diversos enfoques, como bosques aleatorios y redes neuronales recurrentes, se muestran prometedores para predecir los resultados del tratamiento farmacológico y la unión molecular, aunque la precisión varía según la aplicación específica y el conjunto de datos.
Los compuestos generados pueden evaluarse mediante cálculos de campos de fuerza y métricas de propiedades similares a las de los fármacos para determinar su viabilidad.
Aceleración de la ciencia de los materiales
Investigadores de la Universidad Northwestern y del Instituto de Investigación Toyota demostraron el potencial del aprendizaje automático en la síntesis de materiales. Su modelo predijo composiciones de nanomateriales de cuatro, cinco y seis elementos con una característica estructural específica.
¿Los resultados? 18 predicciones correctas de 19 intentos, lo que representa una precisión aproximada del 951 %. Esto no es modelado estadístico; fueron experimentos de síntesis reales que validaron pronósticos computacionales.
| Aplicación de aprendizaje automático | Tasa de precisión | Fuente de datos |
|---|---|---|
| Predicción de la síntesis de nuevos materiales | 95% | 18/19 predicciones correctas |

Aplicar el aprendizaje automático a la investigación química con IA superior
Los proyectos de química suelen basarse en simulaciones, mediciones de laboratorio y conjuntos de datos estructurados que pueden beneficiarse del análisis mediante aprendizaje automático. IA superior Colabora con equipos que exploran el modelado predictivo, el análisis experimental y los flujos de trabajo de investigación asistidos por IA en entornos relacionados con la química.
AI Superior puede brindar soporte a proyectos de química con:
- Análisis de conjuntos de datos experimentales y de simulación
- Desarrollo de modelos de aprendizaje automático para tareas de predicción
- Creación de flujos de trabajo analíticos de prueba de concepto
- Clasificación y reconocimiento de patrones en datos químicos
- Validación del rendimiento y la consistencia del modelo.
- Soporte de integración para sistemas de software de investigación
👉Contacta con AI Superior para analizar el flujo de trabajo previsto.
La realidad del procesamiento de datos
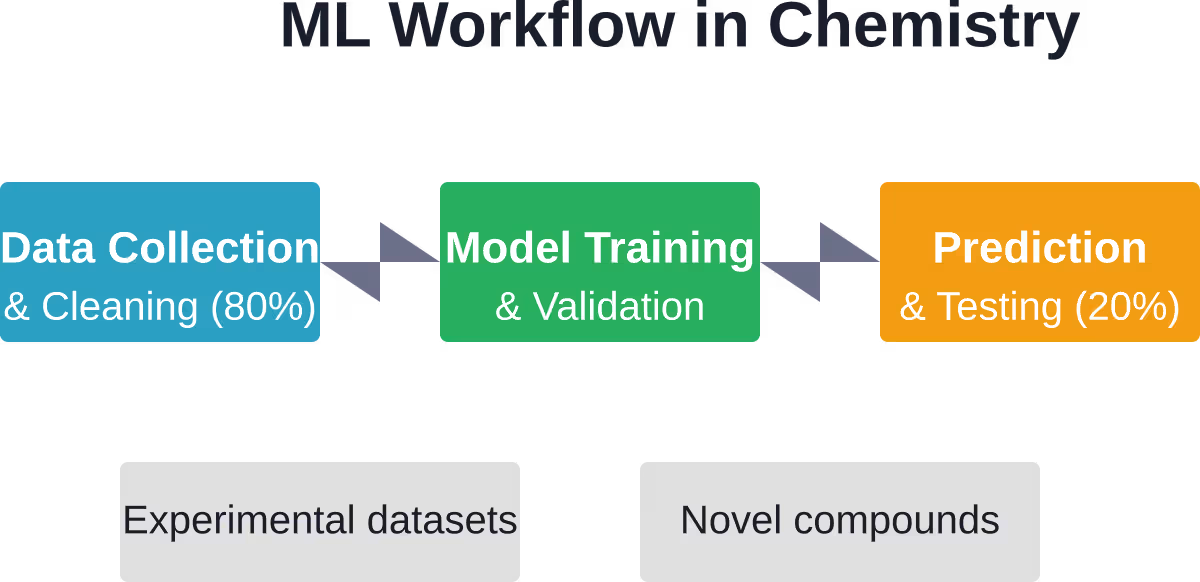
En realidad, el 801% del tiempo de práctica del aprendizaje automático en química se dedica al procesamiento y la limpieza de datos. Solo el 201% se destina a la aplicación de algoritmos. Los conjuntos de datos químicos suelen llegar desordenados, inconsistentes e incompletos.
Esa proporción frustra a los investigadores que esperan soluciones sencillas y rápidas. Pero refleja la complejidad de la química: las condiciones experimentales varían, las técnicas de medición difieren y los estándares de presentación de informes siguen siendo inconsistentes entre laboratorios y a lo largo de las décadas.
La química cuántica se une al aprendizaje profundo.
La química cuántica ab initio predice las propiedades moleculares resolviendo las ecuaciones de Schrödinger para el movimiento de los electrones. ¿Precisa? Sí. ¿Computacionalmente costosa? Sin duda.
Las capas de aprendizaje profundo ahora aproximan estos cálculos cuánticos con una fracción del costo computacional. Los modelos aprenden de simulaciones cuánticas de alta fidelidad y luego predicen propiedades para nuevas moléculas sin repetir el tratamiento mecánico cuántico completo.
Este enfoque híbrido preserva la precisión al tiempo que permite un cribado de alto rendimiento. Se pueden evaluar miles de moléculas en el tiempo que la química cuántica tradicional tarda en procesar solo unas pocas docenas.
Preguntas frecuentes
¿Qué es el aprendizaje automático en química?
El aprendizaje automático en química aplica algoritmos para predecir propiedades moleculares, diseñar nuevos compuestos y acelerar la investigación. Los modelos aprenden de conjuntos de datos químicos para identificar patrones y realizar predicciones sin necesidad de programación explícita para cada escenario.
¿Qué tan precisas son las predicciones de aprendizaje automático para el descubrimiento de fármacos?
La precisión varía según la aplicación. Diversos modelos muestran distintos niveles de rendimiento para las interacciones proteína-proteína y la generación molecular. Sin embargo, las tasas de éxito en los ensayos clínicos se mantienen en torno al 9,6-121 TP3T, lo que demuestra que las predicciones computacionales no garantizan los resultados clínicos.
¿Puede el aprendizaje automático sustituir los experimentos químicos tradicionales?
No del todo. El aprendizaje automático acelera la generación de hipótesis y prioriza los candidatos para su análisis, pero la validación experimental sigue siendo fundamental. El estudio de materiales de Northwestern logró una precisión de predicción del 95%, pero dichas predicciones aún requerían confirmación mediante síntesis en laboratorio.
¿Qué desafíos en materia de datos existen en las aplicaciones de aprendizaje automático en química?
El procesamiento y la limpieza de datos consumen 80% del tiempo del proyecto. Los conjuntos de datos químicos suelen contener formatos inconsistentes, valores faltantes, variaciones experimentales y unidades de medida incompatibles. La estandarización a lo largo de décadas de investigación y en múltiples laboratorios plantea importantes desafíos.
¿Qué áreas de la química se benefician más del aprendizaje automático?
El descubrimiento de fármacos, la ciencia de los materiales y los cálculos de química cuántica muestran resultados sólidos. El cribado de alto rendimiento, la predicción de propiedades moleculares, la planificación de rutas de síntesis y la predicción de la estructura de proteínas se benefician de los enfoques de aprendizaje automático cuando se dispone de datos de calidad suficiente.
¿Qué habilidades necesitan los químicos para utilizar el aprendizaje automático?
Conocimientos básicos de programación (principalmente Python), comprensión de formatos de datos y preprocesamiento, familiaridad con conceptos de aprendizaje automático como la división de datos en conjuntos de entrenamiento y validación, y experiencia en el dominio para interpretar los resultados de forma crítica. La alfabetización en datos es más importante que las matemáticas avanzadas para la mayoría de las aplicaciones.
¿Cómo se integra la química cuántica con el aprendizaje automático?
Los modelos de aprendizaje automático aprenden de costosos cálculos de mecánica cuántica para aproximar resultados con un menor coste computacional. Esto permite la predicción de propiedades a gran escala, manteniendo una precisión a nivel cuántico para sistemas moleculares donde los cálculos ab initio completos serían prohibitivamente lentos.
El aprendizaje automático aún no ha resuelto los grandes desafíos de la química. Sin embargo, la trayectoria es clara: los algoritmos complementan la experiencia humana, aceleran los plazos de descubrimiento y revelan patrones ocultos en décadas de datos experimentales. La precisión en la predicción de materiales del 95% representa un progreso real, no una simple exageración.
Para los investigadores y las organizaciones que exploran estas herramientas, el mensaje es pragmático: invertir fuertemente en infraestructura de datos, mantener expectativas realistas sobre la traslación clínica y recordar que gran parte del trabajo se realiza antes de que se ejecute cualquier algoritmo. La revolución computacional en química premia la preparación minuciosa más que la sofisticación algorítmica.