Korte samenvatting: Machine learning zorgt voor een revolutie in de chemie door de ontdekking van nieuwe geneesmiddelen te versnellen, moleculaire eigenschappen te voorspellen en nieuwe materialen te ontwerpen. Met algoritmes die veelbelovend zijn in het voorspellen van eiwitinteracties en materiaalsynthese, transformeert ML traditioneel chemisch onderzoek van vallen en opstaan naar datagestuurde precisie, waardoor de ontwikkeltijd en -kosten drastisch worden verlaagd.
De farmaceutische industrie wordt geconfronteerd met een ontnuchterende realiteit: het succespercentage van geneesmiddelenontwikkeling schommelt rond de 9,6-121% van fase I-onderzoeken tot uiteindelijke goedkeuring. Traditionele methoden kosten jaren en miljarden dollars, maar mislukken vaker dan dat ze slagen.
Machine learning verandert die situatie. Door enorme chemische datasets te verwerken en patronen te identificeren die voor menselijke onderzoekers onzichtbaar zijn, versnellen deze algoritmen de ontdekkingsprocessen en verbeteren ze de nauwkeurigheid op diverse gebieden.
Geneesmiddelenontwikkeling krijgt een datagestuurde transformatie.
Het punt is echter dat machine learning juist uitblinkt waar traditionele chemie het meest tekortschiet. Patroonherkenning in enorme moleculaire bibliotheken, voorspelling van eigenschappen zonder fysieke synthese en doelwitidentificatie profiteren allemaal van algoritmische precisie.
Deep learning-modellen voorspellen tegenwoordig eiwit-eiwitinteracties met opmerkelijke nauwkeurigheid. Maar de ontwikkeling van geneesmiddelen blijft een uitdaging. Het algehele succespercentage van fase I klinische studies tot goedkeuring van een geneesmiddel ligt rond de 9,6–121 TP3T, hoewel dit aanzienlijk varieert per therapeutisch gebied (bijvoorbeeld ~31 TP3T voor oncologie). De kloof tussen de belofte van silico-modellen en de klinische realiteit blijft aanzienlijk.
Moleculaire generatie en eigenschapsvoorspelling
Generatieve modellen creëren volledig nieuwe moleculaire structuren met de gewenste eigenschappen. Verschillende generatieve benaderingen vertonen verschillende validiteitspercentages voor moleculaire generatie. Dit zijn geen triviale prestaties: het genereren van chemisch plausibele structuren vereist inzicht in bindingsregels, stabiliteitsbeperkingen en synthetische toegankelijkheid.
Machine learning-modellen die gebruikmaken van verschillende benaderingen, zoals random forests en terugkerende neurale netwerken, zijn veelbelovend voor het voorspellen van de uitkomst van medicamenteuze behandelingen en moleculaire binding, hoewel de nauwkeurigheid varieert afhankelijk van de specifieke toepassing en dataset.
De gegenereerde verbindingen kunnen worden geëvalueerd aan de hand van krachtveldberekeningen en meetwaarden voor geneesmiddelachtige eigenschappen om hun levensvatbaarheid te beoordelen.
Versnelling van materiaalkunde
Onderzoekers van Northwestern University en het Toyota Research Institute hebben de kracht van machinaal leren bij de synthese van materialen aangetoond. Hun model voorspelde de samenstelling van nanomaterialen met vier, vijf en zes elementen en een specifieke structurele eigenschap.
Het resultaat? 18 correcte voorspellingen van de 19 pogingen – een nauwkeurigheid van ongeveer 95%. Dit is geen statistische modellering; het waren daadwerkelijke synthese-experimenten die de computationele voorspellingen valideerden.
| ML-toepassing | Nauwkeurigheidspercentage | Gegevensbron |
|---|---|---|
| Voorspelling van de synthese van nieuwe materialen | 95% | 18/19 correcte voorspellingen |

Pas machine learning toe op chemisch onderzoek met superieure AI-prestaties.
Chemieprojecten zijn vaak gebaseerd op simulaties, laboratoriummetingen en gestructureerde datasets die baat kunnen hebben bij machine learning-analyse. AI Superieur Werkt samen met teams die zich bezighouden met voorspellende modellering, experimentele analyse en AI-ondersteunde onderzoeksworkflows in chemiegerelateerde omgevingen.
AI Superior kan chemische projecten ondersteunen met:
- Analyse van experimentele en simulatiegegevens
- Ontwikkeling van ML-modellen voor voorspellingstaken
- Het opzetten van analytische workflows voor het bewijzen van het concept.
- Classificatie en patroonherkenning in chemische data
- Validatie van de prestaties en consistentie van het model
- Integratieondersteuning voor onderzoekssoftwaresystemen
👉Neem contact op met AI Superior om de geplande workflow te bespreken.
De realiteit van gegevensverwerking
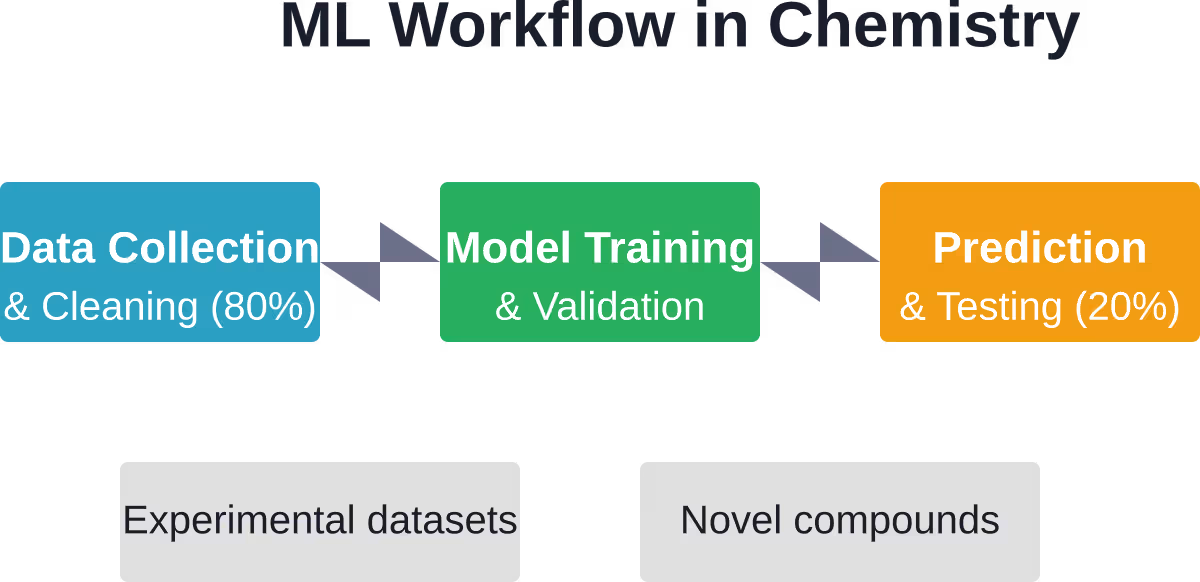
Eerlijk gezegd: 801 TP3T van de machine learning-praktijk in de chemie bestaat uit het verwerken en opschonen van data. Slechts 201 TP3T gaat naar de daadwerkelijke toepassing van algoritmen. Chemische datasets worden vaak rommelig, inconsistent en onvolledig aangeleverd.
Die verhouding frustreert onderzoekers die kant-en-klare oplossingen verwachten. Maar het weerspiegelt de complexiteit van de chemie: experimentele omstandigheden variëren, meettechnieken verschillen en rapportagestandaarden blijven inconsistent tussen laboratoria en decennia.
Kwantumchemie ontmoet diepgaand leren
Ab initio kwantumchemie voorspelt moleculaire eigenschappen door de Schrödinger-vergelijkingen voor elektronenbeweging op te lossen. Nauwkeurig? Jazeker. Rekenkundig kostbaar? Absoluut.
Diepgaande leerlagen benaderen deze kwantumberekeningen nu tegen een fractie van de rekenkosten. Modellen leren van zeer nauwkeurige kwantumsimulaties en voorspellen vervolgens eigenschappen voor nieuwe moleculen zonder de volledige kwantummechanische behandeling te hoeven herhalen.
Deze hybride aanpak behoudt de nauwkeurigheid en maakt tegelijkertijd screening met hoge doorvoer mogelijk. Duizenden moleculen kunnen worden geëvalueerd in de tijd die traditionele kwantumchemie nodig heeft voor tientallen.
Veelgestelde vragen
Wat is machinaal leren in de chemie?
Machine learning in de chemie past algoritmen toe om moleculaire eigenschappen te voorspellen, nieuwe verbindingen te ontwerpen en onderzoek te versnellen. Modellen leren van chemische datasets om patronen te herkennen en voorspellingen te doen zonder expliciete programmering voor elk scenario.
Hoe nauwkeurig zijn ML-voorspellingen voor de ontwikkeling van nieuwe geneesmiddelen?
De nauwkeurigheid varieert per toepassing. Verschillende modellen laten verschillende prestatieniveaus zien voor eiwit-eiwitinteracties en moleculaire generatie. De succespercentages in klinische studies blijven echter rond de 9,6-12% liggen, wat aantoont dat computationele voorspellingen geen garantie bieden voor klinische resultaten.
Kan machinaal leren traditionele scheikunde-experimenten vervangen?
Niet helemaal. Machine learning versnelt het genereren van hypothesen en geeft prioriteit aan kandidaten voor testen, maar experimentele validatie blijft essentieel. De materiaalstudie van Northwestern University behaalde een voorspellingsnauwkeurigheid van 95%, maar die voorspellingen vereisten nog steeds bevestiging door middel van laboratoriumsynthese.
Welke data-uitdagingen bestaan er bij machine learning-toepassingen in de chemie?
Gegevensverwerking en -opschoning nemen 801 TP3T aan projecttijd in beslag. Chemische datasets bevatten vaak inconsistente formaten, ontbrekende waarden, experimentele variaties en incompatibele meeteenheden. Standaardisatie over decennia van onderzoek en meerdere laboratoria vormt een aanzienlijke uitdaging.
Welke vakgebieden binnen de chemie profiteren het meest van machinaal leren?
Geneesmiddelenonderzoek, materiaalkunde en kwantumchemische berekeningen laten sterke resultaten zien. High-throughput screening, voorspelling van moleculaire eigenschappen, planning van syntheseroutes en voorspelling van eiwitstructuren profiteren allemaal van machine learning-benaderingen wanneer er voldoende kwalitatieve data beschikbaar zijn.
Welke vaardigheden hebben chemici nodig om machine learning te kunnen gebruiken?
Basiskennis van programmeren (meestal Python), inzicht in dataformaten en dataverwerking, bekendheid met machine learning-concepten zoals trainings- en validatiesplitsingen, en domeinexpertise om resultaten kritisch te interpreteren. Data-geletterdheid is voor de meeste toepassingen belangrijker dan geavanceerde wiskunde.
Hoe kan kwantumchemie worden geïntegreerd met machinaal leren?
ML-modellen leren van kostbare kwantummechanische berekeningen om resultaten te benaderen tegen lagere rekenkosten. Dit maakt het mogelijk om eigenschappen met een hoge doorvoer te voorspellen, terwijl de nauwkeurigheid op kwantumniveau behouden blijft voor moleculaire systemen waar volledige ab initio-berekeningen onbetaalbaar traag zouden zijn.
Machine learning heeft de grote uitdagingen in de chemie nog niet opgelost. Maar de trend is duidelijk: algoritmes vullen menselijke expertise aan, versnellen ontdekkingsprocessen en onthullen patronen die verborgen liggen in decennia aan experimentele data. De nauwkeurigheid van de materiaalvoorspellingen van 95% vertegenwoordigt echte vooruitgang, geen hype.
Voor onderzoekers en organisaties die deze tools verkennen, is de boodschap pragmatisch: investeer fors in data-infrastructuur, houd realistische verwachtingen over klinische toepassing en onthoud dat het grootste deel van het werk al gedaan is voordat er ook maar één algoritme wordt uitgevoerd. De computationele revolutie in de chemie beloont zorgvuldige voorbereiding meer dan geavanceerde algoritmes.