Kurzzusammenfassung: Maschinelles Lernen in der Genomik nutzt Computer-Algorithmen zur Analyse umfangreicher genetischer Datensätze und identifiziert Muster, die mit traditionellen Methoden nicht erkennbar sind. Von der Vorhersage des Krankheitsrisikos bis zur personalisierten Therapie wandeln ML-Werkzeuge wie Convolutional Neural Networks und überwachte Lernmodelle Rohdaten aus der Genomik in klinische Erkenntnisse um und erzielen in kritischen Anwendungen Leistungsverbesserungen von 7–29% gegenüber herkömmlichen Ansätzen.
Die Genomforschung generiert mehr Daten als je zuvor. Eine einzige Sequenzierung des gesamten Genoms erzeugt Hunderte von Gigabytes. Traditionelle statistische Methoden können da nicht mithalten.
Maschinelles Lernen verändert diese Gleichung. Algorithmen, die mit Millionen von genetischen Varianten trainiert wurden, können Muster erkennen, die Menschen übersehen würden, das Krankheitsrisiko anhand von DNA-Sequenzen vorhersagen und Behandlungsentscheidungen mit beispielloser Präzision steuern.
Laut dem Nationalen Forschungsinstitut für Humangenom (NHGRI) setzen Wissenschaftler zunehmend auf künstliche Intelligenz und maschinelles Lernen, um in komplexen Genomdatensätzen aussagekräftige Muster für die Gesundheitsversorgung und Forschung zu identifizieren. Dieser Wandel ist nicht theoretisch – er findet bereits in Kliniken und Laboren statt.
Warum maschinelles Lernen für die Genomik wichtig ist
Genomdaten sind hochdimensional, verrauscht und so strukturiert, dass herkömmliche Analysemethoden eine Herausforderung darstellen. Ein typisches Exom enthält Varianten in Tausenden von Genen. Die Exomsequenzierung (WES) erfasst etwa 31 T³T des gesamten Genoms, das die Grundlage für proteinkodierende Gene bildet – doch selbst diese 31 T³T erzeugen enorme Datensätze mit Big-Data-Charakteristika.
Maschinelles Lernen ist genau unter diesen Bedingungen besonders effektiv. Wo traditionelle statistische Tests mit Tausenden korrelierter Variablen Schwierigkeiten haben, zeichnen sich ML-Algorithmen durch Folgendes aus:
- Identifizierung nichtlinearer Beziehungen zwischen genetischen Varianten und Phänotypen
- Umgang mit fehlenden Daten und technischem Rauschen, das bei der Sequenzierung auftritt
- Integration heterogener Datenquellen (genomische, transkriptomische, klinische Daten)
- Skalierung auf Datensätze mit Millionen von Stichproben
Das Gebiet erweitert kontinuierlich den Einsatz von computergestützten Methoden, um das Verständnis verborgener Muster in großen, komplexen Genomdatensätzen zu verbessern – von der Grundlagenforschung bis zur klinischen Anwendung.
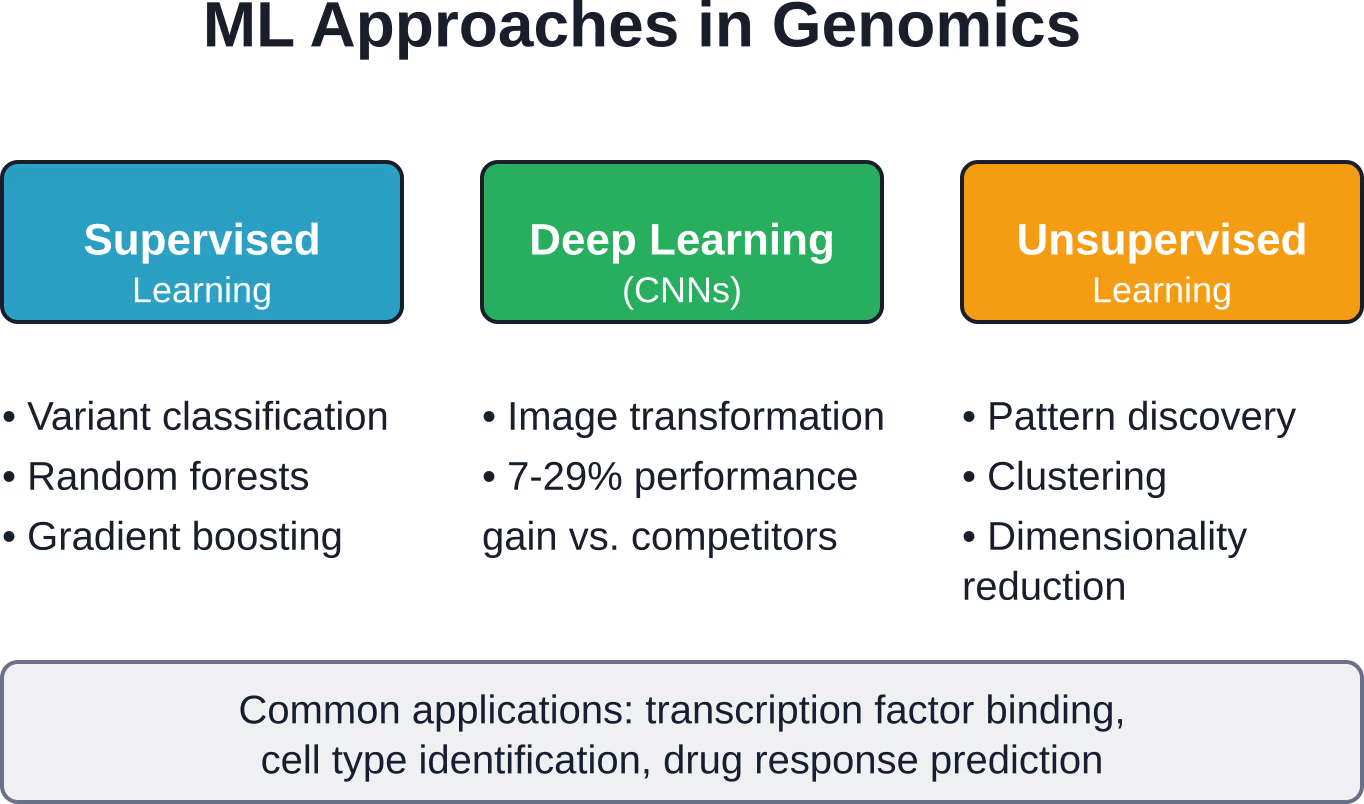
Kernansätze des maschinellen Lernens in der Genomik
Maschinelles Lernen ist nicht gleich maschinelles Lernen. Unterschiedliche genomische Fragestellungen erfordern unterschiedliche algorithmische Strategien.
Überwachtes Lernen zur Variantenklassifizierung
Beim überwachten Lernen werden gelabelte Trainingsdaten verwendet, um Vorhersagemodelle zu erstellen. In der Genomik bedeutet dies, Algorithmen anhand bekannter pathogener und benigner Varianten zu trainieren, um neue, unklare Varianten zu klassifizieren.
Gängige überwachte Techniken umfassen:
- Random Forests, die Entscheidungsbäume kombinieren, um die Pathogenität von Varianten vorherzusagen
- Support-Vektor-Maschinen, die optimale Grenzen zwischen Variantenklassen finden
- Gradient-Boosting-Methoden, die Vorhersagen iterativ verfeinern
Diese Methoden bilden die Grundlage für klinische Variantendatenbanken und Vorhersageinstrumente, die täglich in diagnostischen Laboren eingesetzt werden. Die Genommedizin, die Diagnose- und Behandlungsentscheidungen auf Basis genomischer Variationen ermöglicht, hat Einzug in die klinische Praxis gefunden und ist leichter zugänglich geworden. Die klinische Interpretation der durch Genomanalyse detektierten genomischen Variationen ist in der Genommedizin von entscheidender Bedeutung.
Deep Learning und Convolutional Neural Networks
Deep Learning stellt einen revolutionären Wandel in der prädiktiven Modellierung durch die Anwendung mehrschichtiger neuronaler Netze, insbesondere Convolutional Neural Networks (CNNs), dar.
Das Problem ist jedoch, dass CNNs ursprünglich für die Bildanalyse entwickelt wurden. Forscher entwickelten Transformationsmethoden wie DeepInsight, die Genomdaten von tabellarischer Form in bildähnliche Darstellungen umwandeln und es CNNs so ermöglichen, latente Merkmale effektiv zu erfassen.
Die Ergebnisse sprechen für sich. DeepInsight-3D zeigte laut einer in Nature veröffentlichten Studie eine Leistungssteigerung von 7–29%, gemessen anhand der AUC-ROC-Kurve des Modells, über alle untersuchten Methoden hinweg. DeepInsight-3D erreichte eine durchschnittliche AUC von 0,72 (Fläche unter der Kurve) für die Vorhersage des Ansprechens auf Medikamente.
Transferlernen reduziert die Rechenzeit zusätzlich und verbessert die Leistung. Modelle, die auf großen Genomdatensätzen vortrainiert wurden, können mit kleineren Datensätzen für spezifische Aufgaben feinabgestimmt werden, wodurch die Leistung bei Aufgaben wie der Vorhersage der Transkriptionsfaktorbindung verbessert wird.
Unüberwachtes Lernen zur Mustererkennung
Wenn keine gekennzeichneten Trainingsdaten vorhanden sind, entdeckt unüberwachtes Lernen Strukturen in Genomdaten ohne vordefinierte Kategorien.
Zu den Techniken gehören Clustering-Algorithmen, die ähnliche Proben gruppieren, und Dimensionsreduktionsverfahren, die hochdimensionale Genomdaten in zwei oder drei Dimensionen visualisieren. Diese Ansätze decken verborgene Populationsstrukturen auf, identifizieren Krankheitssubtypen und liefern neue biologische Hypothesen.

Maschinelles Lernen in der Genomforschung mit überlegener KI anwenden
Maschinelles Lernen verändert die Genomik grundlegend, indem es Forschern hilft, riesige genetische Datensätze zu analysieren und aussagekräftige Muster aufzudecken. AI Superior bietet maßgeschneiderte KI- und ML-Lösungen, die auf komplexe Datenherausforderungen in der Genomforschung angewendet werden können.
Nutzen Sie KI für Ihre Genomik-Workflows
AI Superior bietet Funktionen für maschinelles Lernen, die Genomik-Initiativen unterstützen können, wie zum Beispiel:
- Mustererkennung in großen Datenmengen
- Vorhersagemodelle zur Unterstützung bei der Identifizierung von Trends
- Automatisierung von Datenverarbeitungs- und Analyse-Workflows
👉Kontaktieren Sie AI Superior Heute, um zu erfahren, wie ihre KI-Expertise Ihre Genomforschung unterstützen kann.
Anwendungen in der Praxis verändern Forschung und Pflege
Maschinelles Lernen in der Genomik beschränkt sich nicht auf wissenschaftliche Publikationen. Die Anwendungen verändern die klinische Praxis und die biologische Forschung grundlegend.
Vorhersage der Variantenpathogenität
Klinische Variantendatenbanken und maschinelle Lernalgorithmen helfen Ärzten bei der Interpretation der Tausenden von Varianten, die in Patientengenomen entdeckt werden. Mithilfe von Datenbanken wie ClinVar und COSMIC können Tools vorhersagen, ob neu entdeckte Varianten wahrscheinlich eine Krankheit verursachen.
Diese Vorhersagen dienen als Grundlage für diagnostische Entscheidungen, Familienuntersuchungen und die Auswahl der Behandlung bei seltenen genetischen Erkrankungen und Krebs.
Arzneimittelansprechen und Präzisionsonkologie
Multi-Omics-Datenbanken für Krebsforschung, kombiniert mit Modellen des maschinellen Lernens, sagen voraus, wie Tumore auf spezifische Therapien ansprechen werden. Durch die gemeinsame Analyse genomischer, transkriptomischer und proteomischer Daten identifizieren Algorithmen Patienten, die am ehesten von zielgerichteten Behandlungen profitieren.
Die Cancer Cell Line Encyclopedia (CCLE), Genomics of Drug Sensitivity in Cancer (GDSC) und The Cancer Genome Atlas (TCGA) liefern Trainingsdaten für diese Modelle. Forscher haben mit Hilfe von Deep-Learning-Verfahren auf diesen Datensätzen eine Genauigkeit von 72% bei der Vorhersage der Arzneimittelwirksamkeit erzielt.
Transkriptionsfaktorbindung und Genregulation
Das Verständnis der DNA-Bindungsstellen von Transkriptionsfaktoren ist grundlegend für die Entschlüsselung der Genregulation. Maschinelle Lernmodelle, die mit ChIP-seq- und DNase-seq-Daten trainiert wurden, sagen Bindungsstellen allein anhand der DNA-Sequenz voraus.
Das Kipoi-Repository beschleunigt den gemeinschaftlichen Austausch und die Wiederverwendung von Vorhersagemodellen für die Genomik und beherbergt Modelle für die Bindung von Transkriptionsfaktoren, RNA-Splicing und Chromatinzugänglichkeit. Dieser kollaborative Ansatz verhindert redundante Modellentwicklung und ermöglicht systematisches Benchmarking.
Zelltypidentifizierung aus Einzelzelldaten
Die Einzelzell-RNA-Sequenzierung erzeugt Expressionsprofile für Tausende einzelner Zellen. Maschinelles Lernen automatisiert die Zelltypklassifizierung und ersetzt die manuelle Annotation durch skalierbare, reproduzierbare Algorithmen.
Laut veröffentlichten Forschungsergebnissen haben Methoden, die Deep-Learning-Ansätze anwenden, Leistungsverbesserungen bei der Zelltypidentifizierung gezeigt.
| Anwendungsgebiet | ML-Methode | Leistungskennzahl | Klinische Auswirkungen |
|---|---|---|---|
| Variantenpathogenität | Random Forests, SVM | AUC 0,85–0,95 | Diagnostische Klassifizierung |
| TF-Bindungsstellen | CNNs | 15.1% AUPRC-Gewinn | Regulatorisches Verständnis |
| Zellentyp-ID | scDeepInsight | 7%-Verbesserung | Krankheitssubtypisierung |
Genomische Merkmalsentwicklung und Modelleingaben
Der Erfolg von Modellen des maschinellen Lernens hängt entscheidend davon ab, wie genomische Daten dargestellt und welche Merkmale extrahiert werden.
Umfangreiche Genomanalysen haben prädiktive Muster aufgedeckt, die mit Merkmalen und Lebensweisen von Organismen zusammenhängen. Untersuchungen an 387 Pilzgenomen nutzten Merkmalsgruppen, die von kohlenhydrataktiven Enzymen (CAZymes), Peptidasen, Sekundärmetabolitclustern, Transportern und Transkriptionsfaktoren abgeleitet wurden.
Obwohl die Phylogenie in den meisten Vorhersagen eine wichtige Rolle spielte, verbesserte die Einbeziehung genomischer Daten die Vorhersagegenauigkeit für alle getesteten Lebensweisen und Merkmale. Bei der Vorhersage der obligat biotrophen Lebensweise erreichten phylogenetische Daten allein einen AUC-Wert von 0,899 ± 0,018, die Hinzunahme genomischer Merkmale steigerte die Genauigkeit jedoch auf 1,000 ± 0,000, was eine deutliche Verbesserung durch die Integration genomischer Merkmale belegt.
Mal ehrlich: Die Auswahl der Merkmale macht oft den Unterschied zwischen einem mittelmäßigen und einem bahnbrechenden Modell aus.
Hauptmerkmalskategorien
- Sequenzbasierte Merkmale: K-Mer-Frequenzen, GC-Gehalt, Motivhäufigkeiten
- Funktionale Annotationen: Gene-Ontologie-Begriffe, Pathway-Zugehörigkeiten, Proteindomänen
- Evolutionäre Merkmale: Konservierungswerte, phylogenetische Signale, Homologiebeziehungen
- Strukturelle Merkmale: Sekundärstrukturvorhersagen, Chromatinzustand, DNA-Form
Bei der Vorhersage von Nekrotrophen in Pilzgenomen betrug der maximale Anstieg des AUC-Werts 0,395 bei Verwendung des CAZyme-Feature-Sets – ein durchschnittlicher AUC-Gewinn von 87% über die drei besten Feature-Sets im Vergleich zu Sparsamkeitsmethoden.
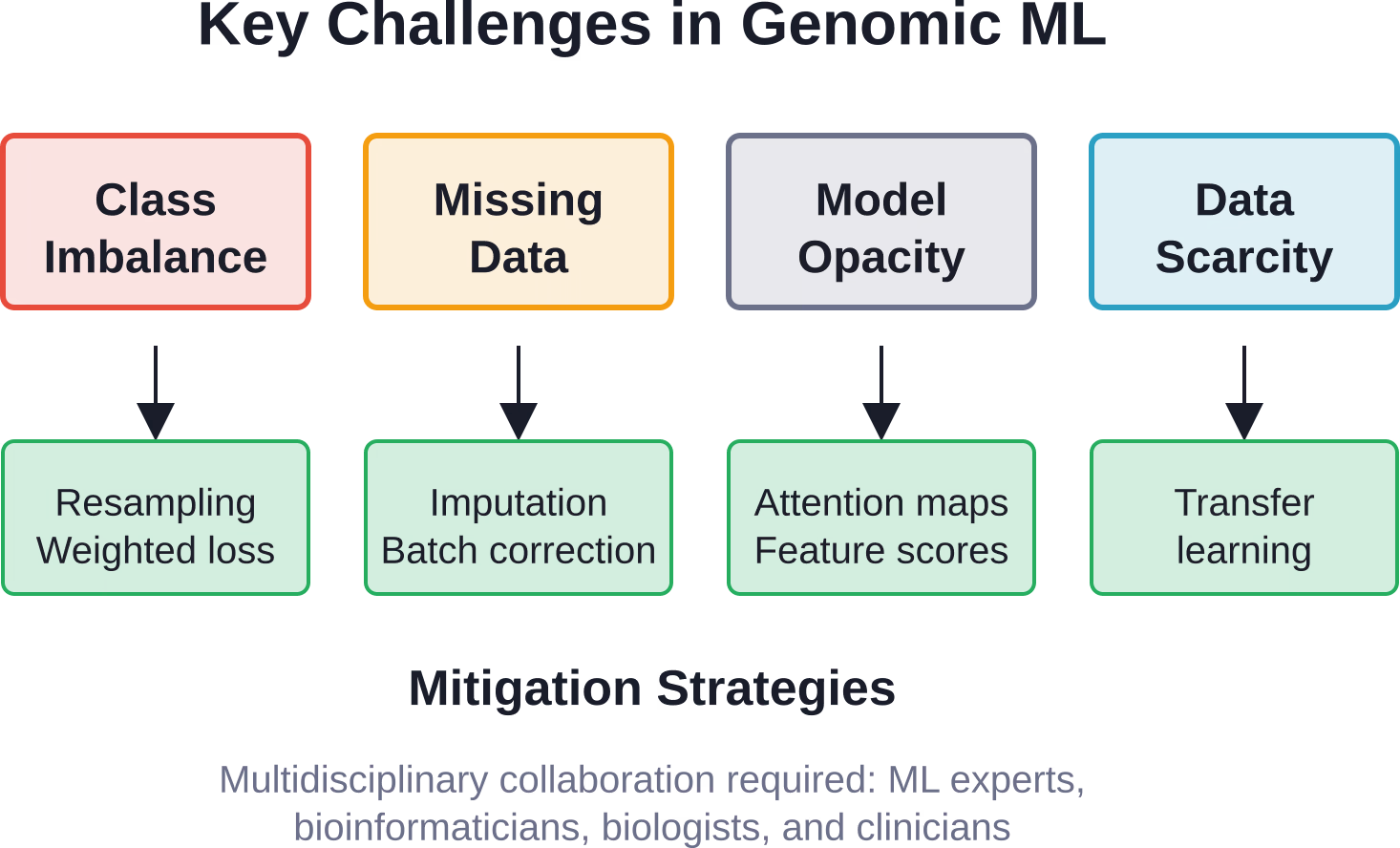
Herausforderungen, die maschinelles Lernen bewältigen muss
Trotz beeindruckender Erfolge steht das maschinelle Lernen in der Genomik vor realen Hindernissen, die die aktuellen Anwendungsmöglichkeiten einschränken.
Ungleiche Klassengrößen
Pathogene Varianten sind im Vergleich zu benignen Varianten selten. Es gibt deutlich weniger Krankheitsfälle als Kontrollfälle. Dieses Ungleichgewicht zwischen den Klassen führt zu einer Verzerrung der Modelle hin zur Mehrheitsklasse und verringert somit die Sensitivität für seltene, klinisch relevante Ereignisse.
Zu den Lösungsansätzen gehören Resampling-Techniken, gewichtete Verlustfunktionen und Ensemble-Methoden, die Ungleichgewichte explizit berücksichtigen.
Fehlende und heterogene Daten
Genomische Datensätze enthalten häufig fehlende Werte aufgrund technischer Fehler, biologischer Abwesenheit oder unvollständiger Datenbanken. Unterschiedliche Sequenzierungsplattformen, Protokolle und Verarbeitungspipelines führen zu Batch-Effekten und Heterogenität.
Fortgeschrittene Imputationsverfahren und Domänenanpassungstechniken sind hilfreich, aber der Umgang mit heterogenen Daten bleibt ein aktives Forschungsgebiet.
Interpretierbarkeit des Modells
Deep-Learning-Modelle sind oft “Black Boxes”. Ein neuronales Netzwerk kann zwar das Krankheitsrisiko genau vorhersagen, liefert aber keine mechanistischen Erkenntnisse darüber, warum eine Variante pathogen ist.
Für die klinische Anwendung ist Interpretierbarkeit entscheidend. Techniken wie Aufmerksamkeitsmechanismen, Saliency-Maps und Feature-Importance-Scores bieten Teillösungen und zeigen auf, welche genomischen Regionen die Vorhersagen beeinflussen.
Datengröße und -qualität
Maschinelles Lernen benötigt viele Daten. Um robuste Modelle zu trainieren, sind Tausende bis Millionen von annotierten Beispielen erforderlich. Für seltene Krankheiten oder wenig erforschte Bevölkerungsgruppen existieren diese Daten schlichtweg noch nicht.
Transferlernen und Few-Shot-Learning-Ansätze zielen darauf ab, aus begrenzten Daten nützliche Modelle zu erstellen, doch die Datenknappheit bleibt eine grundlegende Einschränkung.
Werkzeuge und Ressourcen zur Beschleunigung der Entwicklung
Das Ökosystem der maschinellen Lerngenomik umfasst Repositorien, Datenbanken und kollaborative Rahmenwerke, die die Einstiegshürden senken.
Modell-Repositories
Das Kipoi-Repository stellt vortrainierte Modelle für Genomik-Anwendungen bereit, sodass Forschende bestehende Modelle ohne erneutes Training anwenden können. Dies beschleunigt den Austausch und die Wiederverwendung von Vorhersagemodellen innerhalb der Forschungsgemeinschaft.
Weitere Repositories umfassen:
- MLOmics: Krebs-Multi-Omics-Datenbank, speziell strukturiert für Anwendungen im Bereich maschinelles Lernen
- GitHub-Sammlungen: Von der Community gepflegte Code-Repositories für ML-Workflows in der Genomik
Regierungs- und institutionelle Initiativen
Das Nationale Forschungsinstitut für das Humangenom (NHGRI) hat das Konsortium „ML/AI Tools to Advance Genomic Translational Research“ (MAGen) gegründet. Dieses Forschungsprojekt untersucht die Machbarkeit von Werkzeugen des maschinellen Lernens und der künstlichen Intelligenz, die die Genauigkeit und Präzision der Vorhersage verbessern können, wie sich Krankheiten bei Personen mit pathogenen genetischen Varianten manifestieren.
MAGen bringt das National Institute on Aging (NIA), das Office of Data Science and Strategy (ODSS) und das NHGRI zusammen, um durch koordinierte ML-Entwicklung kritische Fragen der genomischen Translationsforschung anzugehen.
Bildungsressourcen
Kurse und Tutorials helfen Forschern, die notwendigen Programmierkenntnisse zu erwerben, um maschinelles Lernen auf genomische Fragestellungen anzuwenden. Online-Plattformen bieten spezialisierte Kurse zu Genomik und maschinellem Lernen an, und Universitätsstudiengänge integrieren zunehmend computergestützte Genomik in ihre Lehrpläne.
Die Zukunft des maschinellen Lernens in der Genomik
Wie geht es in diesem Bereich weiter? Es zeichnen sich mehrere Trends ab.
Multimodale Integration
Die nächste Generation von Modellen wird Genomsequenzen mit Transkriptom-, Proteom-, Metabolom- und klinischen Daten integrieren. Multi-Omics-Ansätze erfassen die Komplexität biologischer Prozesse umfassender als einzelne Datentypen.
Erste Ergebnisse sind vielversprechend. Modelle, die genomische und transkriptomische Daten kombinieren, übertreffen Ansätze mit nur einer Modalität bei verschiedenen Vorhersageaufgaben.
Grundlagenmodelle für die Genomik
Große Sprachmodelle haben die Verarbeitung natürlicher Sprache revolutioniert. Genomische Fundamentmodelle – massive neuronale Netze, die mit Milliarden von DNA- und RNA-Sequenzen vortrainiert wurden – beginnen, ein ähnliches Potenzial zu zeigen.
Diese Modelle erlernen während des Vortrainings grundlegende Muster der Genombiologie und passen sich anschließend mit minimalen Feinabstimmungsdaten schnell an spezifische Aufgaben an. Dieser Ansatz könnte das genomische maschinelle Lernen demokratisieren, indem er den Datenbedarf für die Entwicklung funktionaler Modelle reduziert.
Methoden zum Schutz der Privatsphäre
Genomische Daten sind von Natur aus sensibel und identifizierbar. Föderiertes Lernen ermöglicht das Training von Modellen über mehrere Institutionen hinweg, ohne die Rohdaten zentral zu speichern. Differenzielle Privatsphäre bietet mathematische Garantien dafür, dass die Modellausgaben keine Informationen auf individueller Ebene preisgeben.
Diese Techniken werden unerlässlich sein, wenn die Genommedizin auf Anwendungen in der gesamten Bevölkerung ausgeweitet wird.
Klinische Entscheidungsunterstützung
Werkzeuge des maschinellen Lernens entwickeln sich von Forschungsprototypen zu FDA-zugelassenen klinischen Entscheidungshilfesystemen. Es ist mit einem weiteren Wachstum der regulatorischen Rahmenbedingungen für genomische KI, standardisierten Leistungsbenchmarks und der Integration in elektronische Patientenakten zu rechnen.
Doch Moment mal. Für die klinische Anwendung ist mehr als nur die technische Leistungsfähigkeit erforderlich. Interpretierbarkeit, Vermeidung von Verzerrungen und Aspekte der Chancengleichheit entscheiden darüber, ob diese Instrumente die Ungleichheiten im Gesundheitswesen verbessern oder verschärfen.
Einstieg in das genomische maschinelle Lernen
Für Forscher, die maschinelles Lernen auf Genomikprobleme anwenden möchten, helfen mehrere praktische Schritte beim Aufbau grundlegender Fähigkeiten:
- Lerne die Biologie: Effektives genomisches maschinelles Lernen erfordert ein Verständnis der biologischen Fragestellungen und der Datengenerierungsprozesse.
- Beherrsche die wichtigsten ML-Techniken: Beginnen Sie mit den Grundlagen des überwachten Lernens, bevor Sie zum Deep Learning übergehen.
- Öffentliche Datensätze erkunden: TCGA, CCLE, GDSC, ClinVar und gnomAD liefern Trainingsdaten für diverse Anwendungen.
- Nutzen Sie etablierte Rahmenbedingungen: Python-Bibliotheken wie scikit-learn, TensorFlow und PyTorch beschleunigen die Entwicklung
- Strenge Benchmarks durchführen: Vergleich neuer Methoden mit etablierten Vergleichsmethoden unter Verwendung zurückgehaltener Testdatensätze
- Disziplinübergreifend zusammenarbeiten: Arbeiten Sie mit Fachexperten zusammen, um die biologische Relevanz und den klinischen Nutzen sicherzustellen.
Von der Community gepflegte Ressourcen wie GitHub-Repositories und Online-Kurse verkürzen die Lernkurve. Das Fachgebiet profitiert von der Open-Source-Kultur und dem Datenaustausch, die schnelle Iterationen ermöglichen.
Häufig gestellte Fragen
Was ist maschinelles Lernen in der Genomik?
Maschinelles Lernen in der Genomik nutzt computergestützte Algorithmen, um genetische Daten zu analysieren, Muster zu erkennen und Vorhersagen über biologische Funktionen, Krankheitsrisiken und Therapieansprechen zu treffen. Diese Methoden bewältigen die hochdimensionale und komplexe Natur genomischer Datensätze effektiver als traditionelle statistische Ansätze.
Wie genau sind Modelle des maschinellen Lernens für die Genomvorhersage?
Die Genauigkeit variiert je nach Anwendung. Klassifikatoren für die Pathogenität von Varianten erreichen AUC-Werte von 0,85–0,95. DeepInsight-Methoden zeigen im Vergleich zu konkurrierenden Ansätzen Leistungsverbesserungen von 7–29%. Die Leistung hängt von der Qualität der Trainingsdaten, dem Feature Engineering und der jeweiligen Vorhersageaufgabe ab.
Was sind die größten Herausforderungen bei der Anwendung von ML in der Genomik?
Zu den zentralen Herausforderungen zählen das Ungleichgewicht zwischen seltenen und häufigen Varianten, fehlende oder heterogene Daten von verschiedenen Sequenzierungsplattformen, die Interpretierbarkeit der Modelle für klinische Entscheidungen sowie begrenzte Trainingsdaten für seltene Erkrankungen oder unterrepräsentierte Bevölkerungsgruppen. Um diese Herausforderungen zu bewältigen, ist eine multidisziplinäre Zusammenarbeit zwischen Experten für maschinelles Lernen, Bioinformatikern und Klinikern erforderlich.
Kann maschinelles Lernen Krankheiten anhand von DNA-Sequenzen vorhersagen?
Maschinelle Lernmodelle können das Krankheitsrisiko anhand genomischer Varianten abschätzen, ihre Vorhersagen sind jedoch probabilistisch und nicht deterministisch. Modelle, die mit großen Datenbanken wie ClinVar trainiert wurden, sagen die Pathogenität von Varianten voraus, um die Diagnose zu unterstützen. Polygenetische Risikoscores kombinieren die Effekte vieler Varianten, um die Krankheitsanfälligkeit abzuschätzen. Umweltfaktoren, Gen-Umwelt-Interaktionen und unvollständiges biologisches Wissen schränken jedoch die Vorhersagegenauigkeit ein.
Worin besteht der Unterschied zwischen überwachtem und unüberwachtem Lernen in der Genomik?
Überwachtes Lernen nutzt gelabelte Trainingsdaten – beispielsweise als pathogen oder gutartig gekennzeichnete Varianten –, um Vorhersagemodelle zu erstellen. Es wird für Klassifizierungs- und Regressionsaufgaben eingesetzt. Unüberwachtes Lernen entdeckt Muster in ungelabelten Daten durch Clustering und Dimensionsreduktion und deckt so Populationsstrukturen oder Krankheitssubtypen ohne vordefinierte Kategorien auf.
Wie verbessert Deep Learning die Genomanalyse?
Deep Learning, insbesondere Convolutional Neural Networks (CNNs), lernt automatisch hierarchische Merkmale aus Rohdaten. Methoden wie DeepInsight transformieren tabellarische Genomdaten in bildähnliche Darstellungen und ermöglichen es CNNs so, komplexe nichtlineare Zusammenhänge zu erfassen. Transfer Learning erlaubt es, auf großen Datensätzen vortrainierte Modelle für spezifische Aufgaben zu optimieren und so die Leistung mit weniger Daten und Rechenaufwand zu verbessern.
Welche Ressourcen gibt es zum Erlernen von genomischem maschinellem Lernen?
Das Kipoi-Repository stellt vortrainierte Modelle und Code bereit. Das MAGen-Konsortium des NHGRI entwickelt kollaborative ML-Tools. Online-Kurse vermitteln die Grundlagen des genomischen maschinellen Lernens. Öffentliche Datenbanken (TCGA, CCLE, GDSC, ClinVar) liefern Trainingsdaten. Python-Bibliotheken (scikit-learn, TensorFlow, PyTorch) bieten Implementierungsframeworks. GitHub-Repositories enthalten von der Community entwickelte Workflows und Tutorials.
Schlussfolgerung
Maschinelles Lernen verändert grundlegend, wie Forscher und Kliniker aus Genomdaten Erkenntnisse gewinnen. Von der Vorhersage der Pathogenität von Varianten bis hin zur personalisierten Krebstherapie liefern ML-Algorithmen Einblicke, die mit traditionellen Methoden unmöglich sind.
Die Leistungssteigerungen sind messbar – 7–29%-Verbesserungen in der Modellgenauigkeit und perfekte AUC-Werte für bestimmte Klassifizierungsaufgaben. Dies sind keine inkrementellen Fortschritte, sondern Quantensprünge in der Leistungsfähigkeit.
Es bestehen weiterhin Herausforderungen. Datenknappheit, Modellinterpretierbarkeit und gerechter Zugang erfordern ständige Aufmerksamkeit. Doch die Entwicklung ist klar: Maschinelles Lernen wird für die Genomik genauso grundlegend werden wie die Sequenzierung selbst.
Für Forschende ist es jetzt an der Zeit, Kompetenzen im Bereich maschinelles Lernen zu entwickeln. Für Kliniker wird das Verständnis dieser Werkzeuge zunehmend unerlässlich für eine evidenzbasierte Praxis. Die Genomforschung erweitert kontinuierlich ihre computergestützten Methoden, um verborgene Muster besser zu verstehen – und diese Muster beginnen sich erst jetzt abzuzeichnen.
Sind Sie bereit, maschinelles Lernen in Ihrer Genomforschung einzusetzen? Beginnen Sie mit öffentlichen Datensätzen, nutzen Sie vortrainierte Modelle aus Repositorien wie Kipoi und arbeiten Sie mit Experten für computergestütztes Lernen zusammen, um die biologische Relevanz und den klinischen Nutzen sicherzustellen.