Kurzzusammenfassung: Maschinelles Lernen in Medizinprodukten nutzt KI-Algorithmen zur Analyse von Gesundheitsdaten, zur Verbesserung der Diagnosegenauigkeit und zur Optimierung der Patientenergebnisse. Die FDA hat 2024 168 ML-fähige Medizinprodukte der Klasse II zugelassen, davon 74,41 TP3T im Bereich Radiologie. Die Zulassung erfolgte auf Grundlage umfassender regulatorischer Rahmenbedingungen, einschließlich der Grundsätze guter Praxis im Bereich maschinelles Lernen (GMLP). Hersteller müssen strenge Validierungsanforderungen, Transparenzstandards und vorgegebene Änderungskontrollpläne erfüllen und gleichzeitig die Sicherheit und Wirksamkeit über den gesamten Produktlebenszyklus hinweg gewährleisten.
Künstliche Intelligenz und maschinelles Lernen verändern das Gesundheitswesen in einem beispiellosen Tempo. Diese Softwarealgorithmen lernen aus der realen Anwendung und verbessern die Geräteleistung kontinuierlich, indem sie aus den täglich im Gesundheitswesen anfallenden riesigen Datenmengen wichtige Erkenntnisse gewinnen.
Sie stellen aber auch besondere Herausforderungen dar. Die Komplexität und der iterative, datengetriebene Charakter der ML-Entwicklung erfordern neue regulatorische Ansätze und Best Practices, für die traditionelle Rahmenbedingungen für Medizinprodukte nicht ausgelegt sind.
Es steht viel auf dem Spiel. Hersteller medizinischer Geräte wetteifern darum, ihre Produkte mit KI-Funktionen auszustatten, um medizinische Fachkräfte besser zu unterstützen und die Patientenversorgung zu verbessern. Allein im Jahr 2024 genehmigte die FDA 168 ML-fähige Medizinprodukte der Klasse II, wobei die Radiologie mit 74,41 Tsd. Tsd. Zulassungen den größten Anteil ausmachte.
Der aktuelle Stand ML-fähiger Medizinprodukte
Die Medizinproduktebranche hat sich dramatisch in Richtung der Integration von maschinellem Lernen gewandelt. Laut FDA-Daten hat die Zulassungsbehörde im Jahr 2024 168 ML-fähige Medizinprodukte der Klasse II zugelassen und damit die über 1.000 KI-gestützten Medizinprodukte ergänzt, die bereits über etablierte Zulassungsverfahren zugelassen wurden.
Die Zulassungsdaten von 2024 geben folgende Aufschlüsse über den aktuellen Markt:
| Metrisch | Prozentualer Wert |
|---|---|
| 510(k)-Vorabmitteilungsweg | 94.6% |
| De-Novo-Klassifizierungspfad | 5.4% |
| Spezialgeräte für die Radiologie | 74.4% |
| Kardiovaskuläre Spezialgeräte | 6.5% |
| Spezialgeräte für die Neurologie | 6.0% |
| Nicht-US-Sponsoren | 57.7% |
| Mittlere Bearbeitungszeit bei der FDA | 162 Tage |
Die Dominanz radiologischer Anwendungen ist nicht überraschend. Medizinische Bildgebung erzeugt enorme Datensätze, die sich hervorragend für maschinelle Lernalgorithmen eignen, um diese zu analysieren, Muster zu erkennen und Anomalien zu identifizieren, die dem menschlichen Auge möglicherweise entgehen.
Anwendungen in der Kardiologie und Neurologie gewinnen jedoch zunehmend an Bedeutung und machen 6,51 TP3T bzw. 6,01 TP3T der Zulassungen im Jahr 2024 aus. Diese Fachgebiete nutzen maschinelles Lernen für Aufgaben wie die EKG-Interpretation, die Schlaganfallerkennung und die Vorhersage von Krampfanfällen.
Die Daten zum Zulassungsverfahren zeigen, dass 94,61 TP3T ML-fähige Medizinprodukte über das 510(k)-Verfahren zugelassen wurden und damit eine weitgehende Gleichwertigkeit mit bestehenden Vergleichsprodukten aufweisen. Lediglich 5,41 TP3T erforderten das De-Novo-Klassifizierungsverfahren für neuartige Medizinprodukte ohne geeignete Vergleichsprodukte.
Internationale Entwicklungstrends
Nicht-US-amerikanische Sponsoren trugen 2024 mit 57,71 Tsd. Billionen US-Dollar zum Erfolg der Zulassungen von ML-gestützten Medizinprodukten bei, was die globale Ausrichtung der Innovationen im Bereich Medizintechnik widerspiegelt. Unternehmen weltweit investieren massiv in KI-gestützte Gesundheitslösungen und konkurrieren darum, fortschrittliche Diagnose- und Therapieverfahren auf den Markt zu bringen.
Die mittlere Bearbeitungszeit der FDA für ML-fähige Geräte betrug im Jahr 2024 insgesamt 162 Tage. Dies bietet den Herstellern einen angemessenen Zeitrahmen für den Markteintritt, obwohl der De-Novo-Weg deutlich mehr Zeit für die Bewertung neuartiger Geräte erfordert.
Maschinelles Lernen in medizinischen Geräteanwendungen verstehen
Maschinelles Lernen in Medizingeräten unterscheidet sich grundlegend von herkömmlicher Software. Diese Systeme nutzen Algorithmen, die aus Daten lernen, sich anhand neuer Informationen anpassen und in manchen Fällen ihre Leistung ohne explizite Neuprogrammierung verbessern.
Die Softwarealgorithmen analysieren Muster in riesigen Datensätzen, die während der Gesundheitsversorgung generiert werden. Sie identifizieren Korrelationen, treffen Vorhersagen und unterstützen klinische Entscheidungen auf eine Weise, die statische, regelbasierte Systeme nicht leisten können.
Mal ehrlich: Diese Anpassungsfähigkeit birgt sowohl Chancen als auch regulatorische Herausforderungen. Ein Gerät, das sein Verhalten auf Basis von Daten nach der Markteinführung ändert, erfordert eine andere Überwachung als ein statisches Gerät mit fester Funktionalität.
Gängige Anwendungen von ML-Geräten
Hersteller von Medizinprodukten setzen maschinelles Lernen in verschiedenen Bereichen des Gesundheitswesens ein:
- Diagnostische Bildanalyse: Erkennung von Tumoren, Frakturen oder anderen Anomalien in Röntgenbildern, MRT-Aufnahmen, CT-Scans und Ultraschallbildern
- Klinische Entscheidungsunterstützung: Empfehlung von Behandlungsoptionen auf Grundlage von Patientendaten, Krankengeschichte und Ergebnisstudien
- Patientenüberwachung: Frühwarnzeichen für eine Verschlechterung des Zustands auf Intensivstationen oder in Fernüberwachungsszenarien erkennen
- Risikostratifizierung: Vorhersage, welche Patienten ein höheres Risiko für bestimmte Erkrankungen oder Komplikationen aufweisen
- Personalisierung der Behandlung: Anpassung der Therapieparameter an die individuellen Patientenmerkmale und -reaktionen
- Workflow-Optimierung: Optimierung klinischer Abläufe, Verkürzung von Wartezeiten und Verbesserung der Ressourcenzuweisung
Jede Anwendung erfordert eine sorgfältige Validierung, um sicherzustellen, dass der ML-Algorithmus bei unterschiedlichen Patientenpopulationen und klinischen Umgebungen sicher und effektiv funktioniert.

Unterstützen Sie die Entwicklung medizinischer Geräte mit überlegener KI und maschinellem Lernen.
Maschinelles Lernen spielt eine zunehmend wichtige Rolle bei der Entwicklung medizinischer Geräte, indem es die Dateninterpretation, die Qualitätskontrolle und die Vorhersagekraft verbessert. AI Superior bietet maßgeschneiderte KI- und ML-Lösungen, die Unternehmen dabei helfen, komplexe Datenherausforderungen zu bewältigen und analytische Arbeitsabläufe zu verbessern.
Setzen Sie KI in Ihren Medizinprodukteprojekten ein
AI Superior unterstützt Anwendungen des maschinellen Lernens, wie zum Beispiel:
- Erweiterte Datenanalyse und Mustererkennung
- Vorhersagemodellierung für Trend- und Leistungseinblicke
- Automatisierung von Datenworkflows und Qualitätskontrollprozessen
👉Kontaktieren Sie AI Superior heute, um zu erfahren, wie deren KI-Expertise Ihre Initiativen im Bereich Medizintechnik unterstützen kann.
Regulierungsrahmen der FDA für ML-Medizinprodukte
Die FDA hat einen umfassenden Regulierungsansatz speziell für KI- und maschinelle Lerntechnologien in Medizinprodukten entwickelt. Dieser Rahmen erkennt an, dass ML-fähige Geräte eine andere Aufsicht erfordern als herkömmliche statische Software.
Am 7. Januar 2025 veröffentlichte die FDA den Entwurf einer Leitlinie: Softwarefunktionen künstlicher Intelligenz für Medizinprodukte: Empfehlungen zum Lebenszyklusmanagement und zur Marktzulassung. Dies ist die erste umfassende Leitlinie, die den gesamten Lebenszyklus von der Entwicklung bis zur Marktbeobachtung abdeckt.
Grundsätze guter maschineller Lernpraxis (GMLP)
Im Oktober 2021 haben Health Canada, die US-amerikanische Food and Drug Administration und die britische Medicines and Healthcare products Regulatory Agency gemeinsam zehn Leitprinzipien für eine gute Praxis im Bereich des maschinellen Lernens festgelegt.
Diese GMLP-Prinzipien unterstützen die Entwicklung sicherer, effektiver und qualitativ hochwertiger KI/ML-Technologien, die aus der realen Anwendung lernen und die Geräteleistung verbessern können. Die Prinzipien berücksichtigen die besonderen Herausforderungen, die sich aus der Komplexität und dem datengetriebenen Entwicklungsansatz von ML ergeben.
Die Leitprinzipien bilden die Grundlage für bewährte Verfahren während des gesamten Produktlebenszyklus, vom ersten Entwurf über die Marktüberwachung bis hin zur kontinuierlichen Verbesserung.
Regulatorische Wege
Medizinprodukte mit maschinellem Lernen gelangen über etablierte regulatorische Wege auf den US-Markt, wobei Anpassungen vorgenommen werden, um ihren besonderen Eigenschaften gerecht zu werden:
- 510(k) Vorabmitteilung: Der dominierende Zulassungsweg für ML-Geräte machte 94,61 TP3T der Zulassungen im Jahr 2024 aus. Die Hersteller wiesen eine weitgehende Gleichwertigkeit mit einem bereits zugelassenen Vergleichsprodukt nach. Die mediane Bearbeitungszeit betrug 2024 151 Tage. Von den 510(k)-zugelassenen ML-fähigen Geräten im Jahr 2024 wiesen 97,51 TP3T identifizierbare Vergleichsprodukte mit einem medianen Alter von 2,2 Jahren auf. Bemerkenswerterweise waren 64,51 TP3T der zitierten Vergleichsprodukte selbst ML-fähige Geräte, was die zunehmende Reife des Ökosystems von KI-gestützten Medizinprodukten widerspiegelt.
- De-novo-Klassifizierung: Für neuartige Medizinprodukte ohne geeignete Vergleichsprodukte bietet der De-Novo-Zulassungsweg Marktzugang für Produkte mit niedrigem bis mittlerem Risiko. Dieser Weg umfasste 5,41 TP3T von 2024 Zulassungen für Medizinprodukte mit einer medianen Bearbeitungszeit von 372 Tagen.
- Vorabzulassung (PMA): Hochrisikofähige, ML-fähige Geräte, die den strengsten Prüfprozess erfordern, werden einer PMA-Bewertung unterzogen, wobei dieser Weg nur einen kleinen Teil der ML-Gerätezulassungen ausmacht.
Vorab festgelegte Änderungskontrollpläne (PCCPs)
Eine der bedeutendsten jüngsten Entwicklungen im Bereich der Regulierung von ML-Geräten ist die Einführung von vorab festgelegten Änderungskontrollplänen. Diese Pläne ermöglichen es Herstellern, spezifische, vorab genehmigte Änderungen an ihren ML-Algorithmen vorzunehmen, ohne dass neue Zulassungsanträge erforderlich sind.
PCCPs begegnen einer entscheidenden Herausforderung: ML-Algorithmen verbessern sich häufig durch erneutes Training mit neuen Daten oder durch algorithmische Verfeinerungen. Traditionelle regulatorische Rahmenbedingungen erforderten für jede Änderung eine neue Einreichung, was zu Engpässen führte und Innovationen verlangsamte.
Im Jahr 2024 enthielten 16,71 TP3T der ML-fähigen Geräte PCCPs in ihren Zusammenfassungen, was die frühe Anwendung dieses neuen regulatorischen Instruments widerspiegelt. Mit zunehmender Erfahrung der Hersteller mit PCCPs und der Präzisierung der Anforderungen durch die Regulierungsbehörden dürfte dieser Anteil steigen.
Bewährte Entwicklungsmethoden für ML-Medizingeräte
Die Entwicklung sicherer und effektiver ML-fähiger Medizinprodukte erfordert strenge, auf die besonderen Eigenschaften von Systemen des maschinellen Lernens zugeschnittene Ingenieurverfahren.
Datenmanagement und -qualität
Maschinelle Lernalgorithmen sind nur so gut wie die Daten, mit denen sie lernen. Datenqualität, Repräsentativität und Diversität beeinflussen die Geräteleistung und potenzielle Verzerrungen direkt.
Zu den bewährten Verfahren gehören:
- Erhebung von Trainingsdaten, die die Zielgruppe hinsichtlich Demografie, Krankheitsbildern und klinischen Umgebungen repräsentieren.
- Dokumentation von Datenquellen, Erhebungsmethoden, Kennzeichnungsverfahren und Qualitätskontrollprozessen
- Implementierung strenger Datenbereinigungs- und Vorverarbeitungsprotokolle
- Validierung der Datenannotationen durch mehrere Experten
- Umgang mit Klassenungleichgewichten und seltenen Fällen in Trainingsdatensätzen
- Aufrechterhaltung der Datenherkunft und Versionskontrolle während der gesamten Entwicklung
Die FDA betont, dass die demografische Vielfalt in den Trainingsdaten unerlässlich ist, um die Leistungsfähigkeit von Medizinprodukten bei verschiedenen Patientengruppen sicherzustellen. Allerdings enthielten nur 15,51 % der ML-fähigen Medizinprodukte demografische Daten in ihren Zulassungsunterlagen für 2024, was eine erhebliche Transparenzlücke offenbart.
Algorithmenentwicklung und -validierung
Die Entwicklung von ML-Algorithmen erfolgt in iterativen Zyklen aus Training, Test, Optimierung und Validierung. Jeder Zyklus muss sorgfältig dokumentiert werden, um die Einreichung bei den Aufsichtsbehörden und die Marktüberwachung zu unterstützen.
Wichtige Überlegungen sind:
- Auswahl geeigneter ML-Architekturen für die klinische Aufgabe (überwachtes Lernen, unüberwachtes Lernen, Deep Learning, Ensemble-Methoden)
- Definition klinisch aussagekräftiger Leistungskennzahlen, die über die reine Genauigkeit hinausgehen
- Verwendung separater Trainings-, Validierungs- und Testdatensätze ohne Überschneidungen
- Durchführung einer externen Validierung von Daten aus verschiedenen Institutionen oder Patientenpopulationen
- Analyse der Algorithmenleistung in verschiedenen demografischen Untergruppen
- Identifizierung und Minderung potenzieller Verzerrungen in algorithmischen Vorhersagen
- Dokumentation aller Hyperparameter, Trainingsverfahren und Modellversionen
Die klinische Validierung muss nachweisen, dass das ML-Gerät in seiner vorgesehenen Einsatzumgebung und mit den vorgesehenen Anwendern sicher und effektiv funktioniert. Laborergebnisse allein reichen nicht aus.
Leistungskennzahlen und Transparenz
Die Definition geeigneter Leistungskennzahlen für ML-basierte Medizinprodukte erfordert klinische Expertise und statistische Strenge. Genauigkeit allein erfasst selten das gesamte klinische Nutzenpotenzial.
Relevante Kennzahlen umfassen häufig Sensitivität, Spezifität, positiven und negativen Vorhersagewert, die Fläche unter der ROC-Kurve und den F1-Score. Die geeigneten Kennzahlen hängen von der klinischen Anwendung und den relativen Kosten falsch positiver gegenüber falsch negativer Ergebnisse ab.
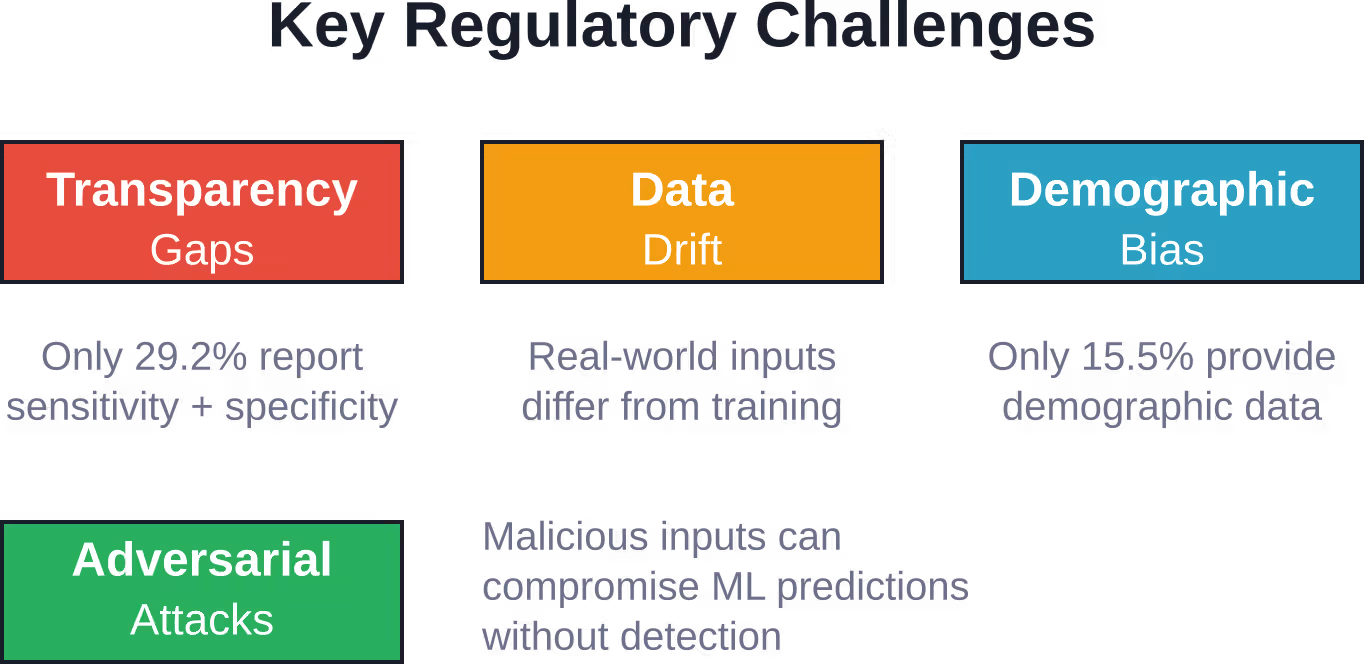
Die Transparenz bei der Leistungsberichterstattung ist jedoch weiterhin uneinheitlich. Laut FDA-Daten aus dem Jahr 2024 berichteten nur 29,21 TP3T der ML-fähigen Geräte in ihren Zulassungsunterlagen sowohl über Sensitivität als auch über Spezifität.
Diese Lücke in der Berichterstattung erschwert die Beurteilung der Leistungsfähigkeit der Geräte und geeigneter Anwendungsfälle durch die Ärzte.
Transparenz- und Berichtspflichten
Transparenz gewährleistet, dass Informationen, die sich auf Patientenrisiken und -ergebnisse auswirken, allen Beteiligten im Umgang mit ML-fähigen Medizinprodukten – Ärzten, Patienten, Gesundheitssystemen und Aufsichtsbehörden – zugänglich gemacht werden.
Wirksame Transparenz bei ML-basierten Medizinprodukten umfasst die Offenlegung von Algorithmusbeschränkungen, Leistungsmerkmalen in verschiedenen Patientengruppen, geeigneten Anwendungsfällen und Kontraindikationen.
Aktuelle Transparenzlücken
Trotz des regulatorischen Fokus auf Transparenz bestehen weiterhin erhebliche Lücken in der FDA-Zulassungsdokumentation für ML-Geräte:
| Transparenzelement | Meldequote (2024) |
|---|---|
| Sowohl Sensitivität als auch Spezifität | 29.2% |
| Demografische Daten | 15.5% |
| Vorab festgelegte Änderungskontrollpläne | 16.7% |
| Überlegungen zur Cybersicherheit | 54.2% |
Diese Lücken erschweren die klinische Entscheidungsfindung. Wie kann ein Radiologe beurteilen, ob ein ML-Algorithmus für seine Patientenpopulation ausreichend funktioniert, ohne demografische Leistungsdaten zu haben? Wie kann ein Krankenhaus Cybersicherheitsrisiken bewerten, ohne klare Sicherheitsinformationen zu erhalten?
Bewährte Verfahren für Transparenz
Führende Hersteller von ML-Medizingeräten implementieren umfassende Transparenzpraktiken:
- Veröffentlichung detaillierter technischer Dokumentationen, die die Algorithmenarchitektur, die Eigenschaften der Trainingsdaten und die Validierungsmethoden beschreiben
- Bereitstellung von Materialien für Klinikpersonal, die geeignete Anwendungsfälle, Leistungskennzahlen und Einschränkungen erläutern.
- Offenlegung der Leistung demografischer Untergruppen zur Identifizierung potenzieller Verzerrungen
- Aktualisierung der Produktkennzeichnung unter Berücksichtigung der Algorithmusänderungen im Rahmen der PCCPs
- Implementierung von Benutzerschnittstellen, die Vertrauensniveaus und Unsicherheiten von Algorithmen kommunizieren.
- Wir bieten Schulungsprogramme an, um die korrekte Verwendung und Interpretation der Geräte sicherzustellen.
Transparenz ist nicht nur eine regulatorische Pflichterfüllung. Sie ist unerlässlich für den Aufbau von Vertrauen bei den Ärzten und die Gewährleistung eines sachgemäßen Einsatzes von Medizinprodukten in der klinischen Praxis.
Marktüberwachung und Leistung im realen Einsatz
Medizinprodukte mit maschinellem Lernen erfordern nach der Marktzulassung eine kontinuierliche Überwachung. Die tatsächliche Leistung kann aufgrund von Unterschieden in der Patientenpopulation, Abweichungen im Arbeitsablauf oder Datenabweichungen von den Ergebnissen kontrollierter Validierungsstudien abweichen.
Datendrift tritt auf, wenn sich die statistischen Eigenschaften der Eingangsdaten im Laufe der Zeit verändern, was die Leistungsfähigkeit von Algorithmen beeinträchtigen kann. Medizinische Bildgebungsprotokolle entwickeln sich weiter, Patientenpopulationen verändern sich und Krankheitsbilder variieren je nach Umgebung.
Überwachungsstrategien
Eine wirksame Marktüberwachung von ML-Geräten umfasst:
- Kontinuierliche Leistungsüberwachung mithilfe von Echtzeitdaten
- Erfassung von unerwünschten Ereignissen und Gerätefehlfunktionen
- Analyse der Leistungsfähigkeit in verschiedenen demografischen Untergruppen und klinischen Umgebungen
- Erkennung von Datenabweichungen durch statistische Überwachung
- Validierung der Algorithmenleistung anhand neuer Datenverteilungen
- Einholen von Nutzerfeedback zur Gerätebenutzerfreundlichkeit und zum klinischen Nutzen
Die Hersteller sollten klare Schwellenwerte für Leistungsverschlechterungen festlegen, die eine Untersuchung und gegebenenfalls ein Nachtraining oder eine Modifizierung des Algorithmus auslösen.
Kontinuierliche Verbesserungszyklen
PCCPs ermöglichen es Herstellern, auf Basis von Daten nach der Markteinführung vorab festgelegte Änderungen ohne neue Zulassungsanträge umzusetzen. Dadurch entsteht ein kontinuierlicher Verbesserungsprozess, in dem Erkenntnisse aus der Praxis die Verfeinerung von Algorithmen vorantreiben.
Kontinuierliche Verbesserungen erfordern jedoch eine sorgfältige Steuerung. Hersteller müssen alle Änderungen dokumentieren, Leistungsverbesserungen nachweisen und die Anwender durch aktualisierte Kennzeichnung und Schulungsmaterialien über die Änderungen informieren.
Das Gleichgewicht zwischen Innovationsgeschwindigkeit und Sicherheitsaufsicht bleibt eine zentrale Herausforderung bei der Regulierung von ML-Geräten. PCCPs stellen einen sich entwickelnden regulatorischen Ansatz zur Bewältigung dieses Gleichgewichts dar.
Überlegungen zur Cybersicherheit
Medizinprodukte mit maschinellem Lernen sind besonderen Cybersicherheitsrisiken ausgesetzt. Diese Geräte verbinden sich häufig mit Krankenhausnetzwerken, übertragen sensible Patientendaten und erhalten möglicherweise Algorithmusaktualisierungen aus der Ferne.
Adversarial Attacks stellen ein besonderes Problem für ML-Systeme dar. Sorgfältig präparierte Eingabedaten können dazu führen, dass Algorithmen falsche Vorhersagen treffen und somit die Patientensicherheit gefährden.
Bewährte Sicherheitspraktiken
Laut Daten aus dem Jahr 2024 haben 54,2% der ML-fähigen Geräte Cybersicherheitsaspekte in ihren Zulassungsdokumenten berücksichtigt – besser als Transparenz in einigen anderen Bereichen, aber immer noch bleibt fast die Hälfte der Geräte mit unklarem Sicherheitsstatus.
Robuste Cybersicherheit für medizinische ML-Geräte umfasst:
- Verschlüsselung von Daten während der Übertragung und im Ruhezustand
- Implementierung sicherer Authentifizierung und Autorisierung
- Protokollierung von Algorithmusänderungen und Benutzerzugriffen
- Validierung von Algorithmusaktualisierungen über sichere Kanäle
- Testen von Algorithmen gegen Angriffe
- Implementierung von Einbruchserkennung und Überwachung
- Aufrechterhaltung des Schwachstellenmanagements und der Patching-Prozesse
Cybersicherheit ist keine einmalige Angelegenheit. Sie erfordert ständige Wachsamkeit, Aktualisierungen zur Abwehr neuer Bedrohungen und die Abstimmung mit den IT-Systemen im Gesundheitswesen.
Internationale Regulierungslandschaft
Die Regulierung von Medizinprodukten für maschinelles Lernen geht über die Zuständigkeit der FDA hinaus. Hersteller, die globale Märkte erschließen wollen, müssen sich in verschiedenen regulatorischen Rahmenbedingungen mit unterschiedlichen Anforderungen zurechtfinden.
europäische Union
Die EU-Medizinprodukteverordnung (MDR) und die Verordnung über In-vitro-Diagnostika (IVDR) regeln Medizinprodukte auf dem europäischen Markt. ML-fähige Geräte fallen unter diese Rahmenbedingungen und werden nach Verwendungszweck und Risikostufe klassifiziert.
Die EU legt Wert auf klinische Evidenz, Marktüberwachung und Transparenz. Einige Anforderungen gehen über die Erwartungen der FDA hinaus, insbesondere hinsichtlich der Dokumentation zur klinischen Validierung.
Internationale Harmonisierungsbemühungen
Die gemeinsame Veröffentlichung der Grundsätze für gute maschinelle Lernpraxis durch Health Canada, die FDA und die britische MHRA im Jahr 2021 stellt einen bedeutenden Fortschritt in Richtung internationaler Harmonisierung dar.
Das International Medical Device Regulators Forum (IMDRF) arbeitet daran, die regulatorischen Ansätze in verschiedenen Rechtsordnungen aufeinander abzustimmen, doppelte Anforderungen zu reduzieren und sichere Innovationen zu beschleunigen.
ISO-Normen für maschinelles Lernen in Medizinprodukten sind in Entwicklung, darunter ISO/DTS 24971-2.2, die eine Anleitung zur Anwendung des Risikomanagements auf ML-fähige Medizinprodukte bietet.
Allerdings bestehen in den verschiedenen Rechtsordnungen weiterhin erhebliche Unterschiede hinsichtlich der Zulassungsfristen, der Anforderungen an klinische Nachweise und der Verpflichtungen nach der Markteinführung.
Anforderungen an die klinische Validierung und den Nachweis
Die klinische Validierung belegt, dass ein ML-basiertes Medizinprodukt in seiner vorgesehenen Anwendungsumgebung sicher und effektiv funktioniert. Laborleistungskennzahlen allein erfassen den klinischen Nutzen in der Praxis nicht.
Die Validierungsanforderungen variieren je nach Risikoklassifizierung des Medizinprodukts, Verwendungszweck und Zulassungsverfahren. Medizinprodukte mit höherem Risiko unterliegen strengeren Nachweisanforderungen.
Überlegungen zum Studiendesign
Aussagekräftige klinische Validierungsstudien für ML-Geräte weisen gemeinsame Merkmale auf:
- Prospektive Datenerhebung, wann immer möglich, um retrospektive Verzerrungen zu vermeiden.
- Mehrere Studienzentren, die unterschiedliche klinische Umgebungen repräsentieren
- Angemessene Stichprobengrößen mit statistischen Powerberechnungen
- Relevante klinische Endpunkte jenseits technischer Leistungskennzahlen
- Vergleich mit dem aktuellen Behandlungsstandard oder der klinischen Praxis
- Analyse der Algorithmenleistung in verschiedenen demografischen Untergruppen
- Unabhängige Validierungsdatensätze, getrennt von den Entwicklungsdaten
- Eine verblindete Auswertung ist, sofern möglich, durchzuführen, um Verzerrungen zu reduzieren.
Eine externe Validierung anhand von Daten von Institutionen, die nicht an der Algorithmenentwicklung beteiligt waren, liefert stärkere Belege für die Generalisierbarkeit als eine interne Validierung allein.
Erkenntnisse aus der Praxis
Randomisierte kontrollierte Studien gelten als Goldstandard für klinische Evidenz, doch werden traditionelle Studiendaten für ML-Geräte zunehmend durch Erkenntnisse aus der Praxis ergänzt.
Daten aus der Praxis stammen aus der klinischen Routine, elektronischen Patientenakten, Registern und der Marktbeobachtung. Sie belegen die Leistungsfähigkeit der Geräte in unterschiedlichen, unkontrollierten Umgebungen, die die tatsächlichen Anwendungsbedingungen besser widerspiegeln.
Die Herausforderung bei Daten aus der realen Welt besteht darin, die Datenqualität sicherzustellen und Störfaktoren zu kontrollieren. Beobachtungsdaten weisen nicht die Strenge kontrollierter Studien auf und erfordern daher eine sorgfältige Analyse, um valide Schlussfolgerungen zu ziehen.
Risikomanagement für ML-Medizinprodukte
ISO 14971 stellt den internationalen Standard für das Risikomanagement von Medizinprodukten dar. Die Anwendung dieses Rahmens auf ML-fähige Geräte erfordert die Berücksichtigung der spezifischen Risiken, die mit adaptiven Algorithmen verbunden sind.
ML-spezifische Risiken
Über die herkömmlichen Geräterisiken hinaus stehen ML-Systeme vor besonderen Herausforderungen:
- Risiken der Datenqualität: Falsche, verzerrte oder nicht repräsentative Trainingsdaten führen zu fehlerhaften Algorithmen.
- Überanpassung: Algorithmen, die bei Trainingsdaten gut funktionieren, aber bei neuen Daten schlecht.
- Datendrift: Sich ändernde Eingangsdatenverteilungen verschlechtern die Leistung des Algorithmus im Laufe der Zeit
- Algorithmische Verzerrung: Systematische Fehler, die bestimmte demografische Gruppen oder klinische Erscheinungsbilder betreffen
- Angriffe von Gegnern: Bösartige Eingaben, die darauf abzielen, falsche Vorhersagen zu verursachen
- Integrationsfehler: Probleme, die durch die Interaktion des Geräts mit klinischen Arbeitsabläufen oder IT-Systemen entstehen
- Missverständnis des Nutzers: Kliniker, die Algorithmusausgaben falsch interpretieren oder Geräte unsachgemäß anwenden
Risikominderungsstrategien müssen diese ML-spezifischen Bedenken durch sorgfältige Konzeption, Validierung, Überwachung und Anwenderschulung berücksichtigen.
Nutzen-Risiko-Bewertung
Die FDA-Zulassungsentscheidungen wägen den Nutzen von Medizinprodukten gegen potenzielle Risiken ab. Bei ML-Medizinprodukten werden dabei sowohl die technische Leistungsfähigkeit als auch der klinische Nutzen berücksichtigt.
Ein hochpräziser Algorithmus, der den klinischen Arbeitsablauf stört oder zu einer Alarmmüdigkeit führt, bietet möglicherweise einen geringeren Nettonutzen als ein mäßig präziser Algorithmus, der gut in die Behandlungsprozesse integriert ist.
Nutzen-Risiko-Profile können je nach klinischem Umfeld, Patientenpopulation und Anwendungsfall variieren. Ein Gerät, das für den spezialisierten Einsatz in akademischen medizinischen Zentren geeignet ist, kann in ressourcenarmen Einrichtungen der ambulanten Versorgung inakzeptable Risiken bergen.
Die Zukunft des maschinellen Lernens in Medizinprodukten
Maschinelles Lernen in Medizingeräten entwickelt sich weiterhin rasant. Mehrere Trends werden das Feld in den kommenden Jahren prägen.
Föderiertes Lernen
Föderiertes Lernen ermöglicht das Training von Algorithmen auf verteilten Datensätzen, ohne Patientendaten zentral zu speichern. Krankenhäuser arbeiten bei der Algorithmenentwicklung zusammen und gewährleisten dabei Datenschutz und Datensicherheit.
Dieser Ansatz überwindet Hürden beim Datenzugang und ermöglicht Schulungen mit größeren und vielfältigeren Datensätzen, als sie einzelne Institutionen bereitstellen könnten. Die regulatorischen Rahmenbedingungen werden angepasst, um föderierte Lernansätze zu fördern.
Erklärbare KI
Black-Box-Algorithmen, die Vorhersagen ohne Erklärung liefern, geben Anlass zur Sorge hinsichtlich ihrer klinischen Anwendung. Erklärbare KI-Methoden zielen darauf ab, die Algorithmen transparent und nachvollziehbar zu machen.
Techniken wie Aufmerksamkeitsmechanismen, Saliency-Maps und Feature-Importance-Analysen helfen Klinikern zu verstehen, welche Faktoren die Vorhersagen von Algorithmen beeinflussen. Diese Transparenz schafft Vertrauen und ermöglicht es Klinikern, potenzielle Fehler zu erkennen.
Multimodales Lernen
Zukünftige medizinische Geräte mit maschinellem Lernen werden zunehmend mehrere Datentypen integrieren – Bildgebung, Laborergebnisse, klinische Befunde, physiologische Überwachung, Genomik –, um umfassendere Erkenntnisse zu gewinnen als Algorithmen, die nur eine Modalität berücksichtigen.
Multimodales Lernen birgt sowohl Chancen als auch Herausforderungen für die Validierung, da die Komplexität integrierter Systeme über einfache Bildgebungs- oder Überwachungsanwendungen hinausgeht.
Edge-Computing
Die Ausführung von ML-Algorithmen auf Edge-Geräten anstatt auf zentralen Servern reduziert die Latenz, verbessert den Datenschutz und ermöglicht Echtzeit-Entscheidungsunterstützung. Der Einsatz am Edge erfordert die Optimierung der Algorithmen für ressourcenbeschränkte Umgebungen.
Die regulatorischen Rahmenbedingungen müssen sich an Edge-Deployment-Modelle anpassen, bei denen Algorithmusaktualisierungen durch verteilte Mechanismen und nicht durch zentrale Steuerung erfolgen.
Praktische Empfehlungen für Hersteller
Organisationen, die ML-fähige Medizinprodukte entwickeln, sollten mehrere wichtige Praktiken priorisieren:
Beginnen Sie mit klar definierten klinischen Bedürfnissen. Die erfolgreichsten ML-Systeme lösen reale klinische Probleme mit nachweisbaren Auswirkungen auf Patientenergebnisse, Arbeitsabläufe oder die Versorgungsqualität.
Investieren Sie in hochwertige, repräsentative Trainingsdaten. Die Datenqualität ist für die Leistungsfähigkeit von Algorithmen wichtiger als deren architektonische Komplexität. Vielfältige Daten beugen demografischen Verzerrungen vor.
Implementieren Sie strenge Validierungsprozesse. Externe Validierung anhand unabhängiger Datensätze liefert aussagekräftigere Ergebnisse als interne Tests allein. Analysieren Sie die Leistung in verschiedenen demografischen Untergruppen.
Dokumentieren Sie alles. Für behördliche Zulassungsanträge ist eine detaillierte Dokumentation der Datenquellen, Schulungsverfahren, Validierungsmethoden und Leistungskennzahlen erforderlich. Etablieren Sie frühzeitig entsprechende Dokumentationspraktiken.
Planen Sie die Überwachung nach der Markteinführung ein. Die Überwachung der Leistung im realen Einsatz und kontinuierliche Verbesserungszyklen sind für den Erfolg von ML-Geräten unerlässlich.
Nehmen Sie frühzeitig Kontakt mit den Aufsichtsbehörden auf. Vorgespräche mit der FDA oder anderen Aufsichtsbehörden klären die Erwartungen und reduzieren die Risiken hinsichtlich des Zulassungsverfahrens.
Transparenz sollte oberste Priorität haben. Die umfassende Offenlegung der Eigenschaften, Grenzen und Leistungsfähigkeit des Algorithmus schafft Vertrauen bei den Ärzten und unterstützt den sachgemäßen Einsatz des Geräts.
Berücksichtigen Sie PCCPs. Vordefinierte Änderungskontrollpläne ermöglichen schnellere Algorithmusverbesserungen auf Basis von Daten nach der Markteinführung. Entwickeln Sie PCCP-Strategien frühzeitig in der Geräteentwicklung.
Häufig gestellte Fragen
Welcher Prozentsatz der von der FDA zugelassenen ML-Medizinprodukte nutzt den 510(k)-Zulassungsweg?
Laut FDA-Daten wurden 94,61 % der im Jahr 2024 zugelassenen Medizinprodukte der Klasse II mit maschinellem Lernen über das 510(k)-Verfahren zugelassen. Lediglich 5,41 % benötigten das De-Novo-Verfahren für neuartige Medizinprodukte ohne geeignete Vergleichsprodukte. Das 510(k)-Verfahren ermöglicht es Herstellern, die wesentliche Gleichwertigkeit mit bereits zugelassenen Vergleichsprodukten nachzuweisen und so die Zulassung für Medizinprodukte, die bereits auf dem Markt befindlichen Produkten mit maschinellem Lernen ähneln, zu beschleunigen.
Wie lange dauert die FDA-Prüfung von ML-fähigen Medizinprodukten?
Die mediane Bearbeitungszeit der FDA für ML-fähige Medizinprodukte betrug im Jahr 2024 insgesamt 162 Tage. Aufgeschlüsselt nach Zulassungsweg: Für 510(k)-Geräte lag die mediane Bearbeitungszeit bei 151 Tagen, während für De-Novo-Geräte mit median 372 Tagen deutlich mehr Zeit benötigt wurde. Die Bearbeitungszeiten variieren je nach Komplexität, Neuheitsgrad, Risikoklassifizierung und Vollständigkeit der ursprünglichen Einreichungen des jeweiligen Medizinprodukts.
Was sind vordefinierte Änderungskontrollpläne (PCCPs) für ML-Geräte?
PCCPs ermöglichen es Herstellern, spezifische, vorab genehmigte Änderungen an ML-Algorithmen vorzunehmen, ohne für jede Änderung einen neuen Zulassungsantrag stellen zu müssen. Dies adressiert eine zentrale Herausforderung adaptiver ML-Systeme, die sich durch Nachtraining oder algorithmische Optimierungen verbessern. Im Jahr 2024 enthielten 16,71 TP3T ML-fähige Medizinprodukte PCCPs in ihrer Zulassungsdokumentation. Hersteller legen in ihren PCCPs die Art der geplanten Änderungen, die Akzeptanzkriterien für Änderungen und die Überwachungsprozesse fest.
In welcher medizinischen Fachrichtung sind die meisten ML-fähigen Geräte zugelassen?
Die Radiologie dominiert die Anwendungen von Medizinprodukten mit maschinellem Lernen und machte 2024 74,41 Tsd. Billionen der von der FDA zugelassenen Geräte aus. Medizinische Bildgebung generiert enorme Datensätze, die sich ideal für maschinelles Lernen eignen. Kardiovaskuläre Anwendungen repräsentierten 6,51 Tsd. Billionen der Zulassungen im Jahr 2024, gefolgt von der Neurologie mit 6,01 Tsd. Billionen. Die Konzentration in der Radiologie spiegelt sowohl den hohen Datenbedarf der Bildgebung als auch die vergleichsweise einfache Validierung von Bildanalysealgorithmen wider.
Was sind die Grundsätze guter maschineller Lernpraxis (GMLP)?
Die GMLP-Prinzipien sind zehn Leitprinzipien, die 2021 von Health Canada, der FDA und der britischen MHRA gemeinsam zur Förderung der Entwicklung sicherer, wirksamer und qualitativ hochwertiger Medizinprodukte für maschinelles Lernen (ML) festgelegt wurden. Diese Prinzipien berücksichtigen die besonderen Herausforderungen der Komplexität und datengetriebenen Entwicklung von ML-Technologien. Sie dienen als Grundlage für bewährte Verfahren entlang des gesamten Produktlebenszyklus, von der Entwicklung bis zur Marktüberwachung, und bilden die Basis für eine internationale Harmonisierung der regulatorischen Vorgaben.
Wie verbreitet ist Transparenzberichterstattung über die Leistung von ML-Geräten?
Die Transparenzberichterstattung für ML-basierte Medizinprodukte weist erhebliche Lücken auf. Im Jahr 2024 berichteten lediglich 29,21 % der zugelassenen Geräte über Sensitivität und Spezifität. Nur 15,51 % lieferten demografische Daten zu Trainings- oder Validierungspopulationen. Diese Lücken erschweren die klinische Bewertung der Geräteleistung und geeigneter Anwendungsfälle. Aufsichtsbehörden und die Forschungsgemeinschaft betonen daher zunehmend die Bedeutung umfassender Transparenz für einen sicheren und effektiven Geräteeinsatz.
Was ist Datendrift und warum ist sie für medizinische ML-Geräte relevant?
Datendrift tritt auf, wenn sich die statistischen Eigenschaften der Eingangsdaten im Laufe der Zeit ändern, was die Leistung von ML-Algorithmen beeinträchtigen kann. Medizinische Bildgebungsprotokolle entwickeln sich weiter, Patientenpopulationen verändern sich, Krankheitsbilder variieren je nach Umgebung und Gerätecharakteristika unterscheiden sich. Ein Algorithmus, der mit einer bestimmten Datenverteilung trainiert wurde, kann schlechte Ergebnisse liefern, wenn die realen Eingangsdaten von den Eigenschaften der Trainingsdaten abweichen. Die Marktüberwachung muss Datendrift durch statistische Analysen erkennen, und Hersteller müssen Algorithmen gegebenenfalls mit aktualisierten Daten neu trainieren, um die Leistung aufrechtzuerhalten.
Schlussfolgerung
Maschinelles Lernen in Medizinprodukten zählt zu den vielversprechendsten technologischen Entwicklungen im Gesundheitswesen. Allein im Jahr 2024 genehmigte die FDA 168 ML-fähige Geräte und ergänzte damit die Hunderte von KI-gestützten Diagnose- und Therapiegeräten, die bereits klinisch im Einsatz sind.
Doch Versprechen bringen Verantwortung mit sich. Diese adaptiven Algorithmen erfordern strenge Entwicklungsmethoden, umfassende Validierung, transparente Leistungsberichterstattung und kontinuierliche Überwachung nach der Markteinführung, um die Patientensicherheit und die klinische Wirksamkeit zu gewährleisten.
Die regulatorischen Rahmenbedingungen entwickeln sich stetig weiter. Rahmenwerke wie die Good Machine Learning Practice Principles, Predetermined Change Control Plans und Total Product Lifecycle Guidelines bieten den Herstellern klarere Wege zur Marktzulassung und gewährleisten gleichzeitig eine angemessene Aufsicht.
Es bestehen weiterhin Transparenzlücken, insbesondere bei der Leistungsberichterstattung über demografische Untergruppen hinweg und bei der Offenlegung der Merkmale von Trainingsdaten. Die Branche muss diese Defizite beheben, um das Vertrauen der Ärzte zu stärken und eine gleichberechtigte Geräteperformance bei unterschiedlichen Patientengruppen zu gewährleisten.
Für Hersteller erfordert Erfolg mehr als ausgefeilte Algorithmen. Hochwertige, repräsentative Daten, strenge Validierung, umfassende Dokumentation und das Engagement für kontinuierliche Verbesserung unterscheiden Geräte, die die Patientenversorgung sinnvoll verbessern, von solchen, die im realen Einsatz Schwierigkeiten haben.
Die nächste Generation von ML-Medizingeräten wird voraussichtlich föderiertes Lernen, erklärbare KI, multimodale Datenintegration und Edge Computing umfassen – was die regulatorischen Rahmenbedingungen erneut dazu zwingen wird, sich an den technologischen Fortschritt anzupassen.
Für Gesundheitsdienstleister ist es unerlässlich, die Fähigkeiten, Grenzen und geeigneten Anwendungsfälle von ML-Geräten zu verstehen. Diese Tools ergänzen die klinische Beurteilung, ersetzen sie aber nicht, und ein effektiver Einsatz erfordert eine durchdachte Integration in die klinischen Arbeitsabläufe.
Die Transformation des Gesundheitswesens durch maschinelles Lernen ist im Gange. Der Erfolg hängt von der Zusammenarbeit zwischen Herstellern, Aufsichtsbehörden, Ärzten und Patienten ab, um sicherzustellen, dass diese leistungsstarken Technologien ihr Potenzial voll ausschöpfen und gleichzeitig die für die medizinische Praxis erforderliche Sicherheit und Wirksamkeit gewährleisten.
Sind Sie bereit, Ihr ML-fähiges Medizinprodukt zu entwickeln? Beginnen Sie mit der Durchsicht der FDA-Leitlinien, der Zusammenstellung hochwertiger und vielfältiger Trainingsdaten und dem frühzeitigen Austausch mit Experten für regulatorische Angelegenheiten in Ihrem Entwicklungsprozess.